Bug Report: Incorrect bond perception for Na-containing xyz files in Avogadro 2
Category: Support / Bug Report
Avogadro version: 2.0.0
OS: Windows 10/11
Related: Bond perception, Na atom, xyz file
Summary
When opening an xyz file containing sodium (Na) atoms in Avogadro 2, the bond structure is severely distorted. The same file displays correctly in Avogadro 1.2.0.
Steps to Reproduce
- Create or obtain an xyz file of a Na-containing organic salt (e.g., disodium phenolphthalein, C₂₀H₁₂Na₂O₄, 38 atoms, charge = 0)
- Open the file in Avogadro 2.0.0
- Observe the 3D structure
Minimal xyz example (disodium phenolphthalein quinoid form):
38
XYZ file generated by Avogadro.
C 1.39816 -0.35703 0.64968
C 0.69037 -1.35396 -0.02097
...
Na (coordinates)
Na (coordinates)
O (coordinates)
...
(Full xyz file available on request)
Expected Behavior
The organic framework (C₂₀H₁₂O₄) should display with correct bond connectivity, similar to Avogadro 1.x behavior. Na⁺ ions should appear as isolated atoms or with correct ionic contacts only.
Actual Behavior
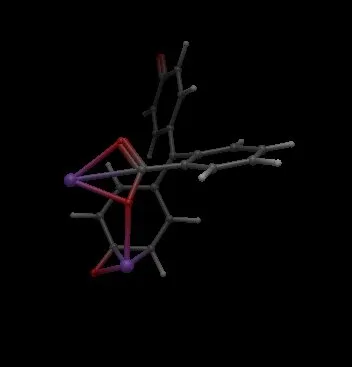
The structure is severely distorted: Na atoms form spurious covalent bonds to neighboring organic atoms, which in turn causes the bond order perception algorithm to remove or misassign C–C and C–O bonds throughout the molecule. The result is an unrecognizable geometry.
Screenshot comparison:
- Avogadro 1.x: Correct structure ✓
- Avogadro 2.0.0: Severely distorted structure ✗
Root Cause Analysis
The issue appears to stem from two compounding factors:
-
Na covalent radius threshold: The Na atom (covalent radius ~1.66 Å) is close enough to neighboring oxygen atoms that the distance-based bond perception algorithm incorrectly assigns covalent bonds to what are actually ionic Na⁺···O⁻ contacts (~2.3–2.5 Å).
-
Valence overflow cascade: Once spurious Na–O bonds are created, the OpenBabel valence-correction step removes legitimate C–C and C–O bonds to satisfy valence rules, collapsing the organic framework.
This is consistent with the known issue reported in the literature (Dominguez et al., J. Chem. Inf. Model. 2020, 60, 3352–3364) where OpenBabel 2.3.x overestimates covalent bonds to alkali metal atoms.
Workaround
Use Avogadro 1.2.0 for visualization of Na-containing xyz files. The ORCA quantum chemistry calculations themselves are unaffected.
Suggested Fix
Consider adding a special case in the bond perception algorithm for alkali metals (Li, Na, K, Rb, Cs):
- Do not assign covalent bonds to Na if the distance exceeds ~2.0 Å (i.e., treat contacts >2.0 Å as ionic, not covalent)
- Or add Na to a list of “ionic-only” atoms that are excluded from the valence-correction cascade
Alternatively, exposing a user-adjustable bond distance threshold per element would allow users to work around this without a code change.
References
- Dominguez et al., J. Chem. Inf. Model. 2020, 60, 3352 — bond perception benchmark showing OpenBabel overestimates metal bonds: https://pubs.acs.org/doi/10.1021/acs.jcim.0c00076
- Related forum thread: Bonds not detected and displayed
- Related forum thread: Bond order issues with xyz files
Thank you for the continued development of Avogadro 2 — it is a valuable tool for the computational chemistry community.
(or via file-sharing service like Dropbox or Pastebin)