Avogadro version: v 1.95.1 (ubunut 22.04 package) and v1.97 (AppImage)
Operating system and version:
Ubuntu 22.04
Expected Behavior
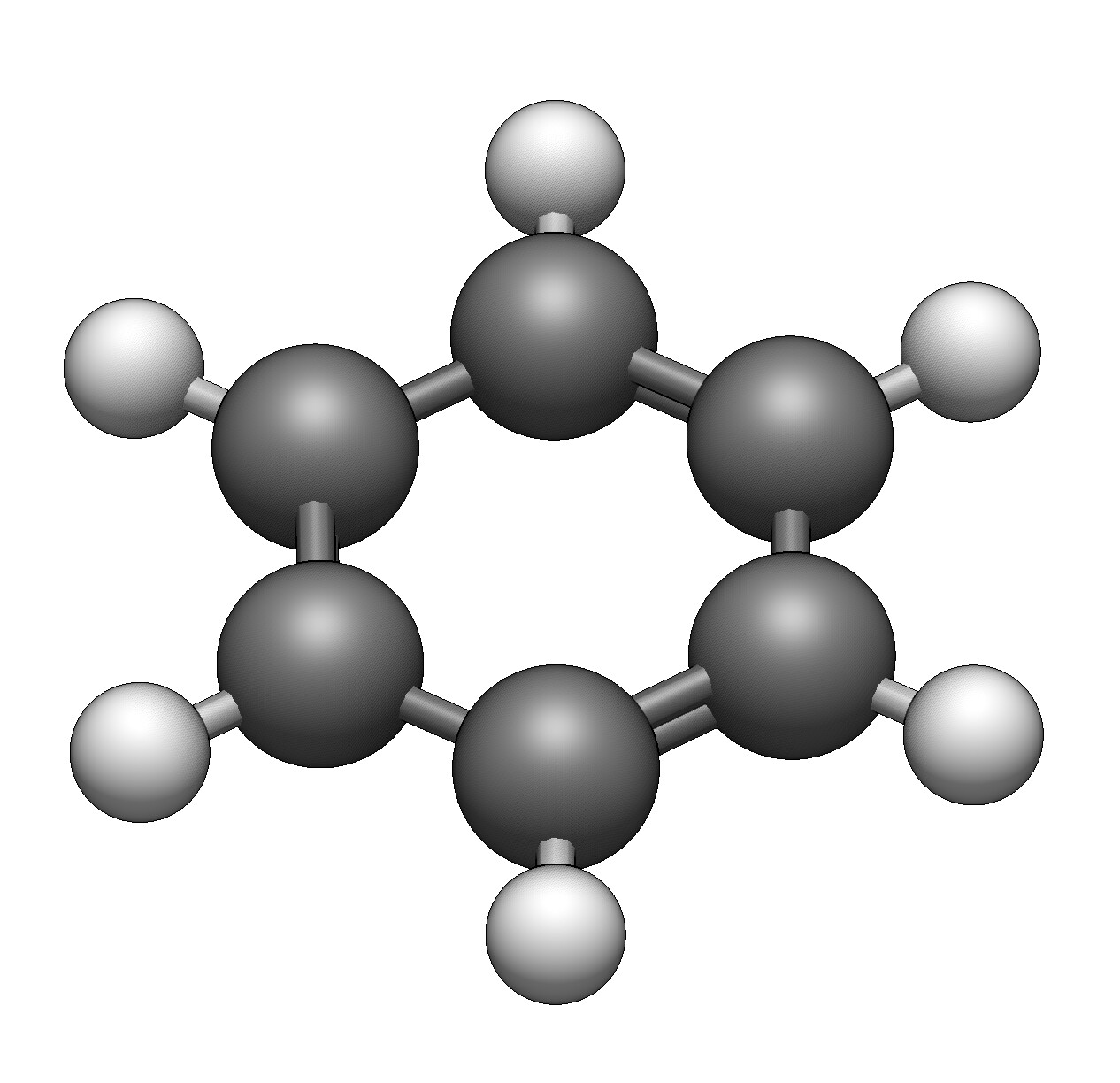
opening an xyz file of a benzene ring and hoping to see the double bonds
Actual Behavior
only single bonds
Steps to Reproduce
save the following data as an xyz, and open it.
12
Coordinates from ORCA-job r2SCAN
C 0.90155 -7.59274 3.38358
C 1.14305 -8.96573 3.38747
C 0.94901 -8.98361 0.98158
C 1.16612 -9.65948 2.18093
C 0.68513 -6.92178 2.18369
H 1.30924 -9.48179 4.30994
C 0.70550 -7.60784 0.96332
H 0.50077 -5.86783 2.19389
H 0.53893 -7.09204 0.04077
H 1.35085 -10.71339 2.17425
H 0.96911 -9.52695 0.06002
H 0.88251 -7.05253 4.30701
Well, an .xyz file does not store information about bond orders, nor about which atom is connected is to an other. One approach to assign/guess bond order is to determine the distance of an atom and its neighbors and to compare this with reference values (e.g., van der Waals radii, or tabulated bond lengths already determined experimentally) with a check what by valence is plausible. xyz2mol is an example of a GUI-less implementation to (re)create a connectivity table seen in .mol and .sdf files, for example.
And second (which once was an omission I forgot to spot), if selecting the representation ball and sticks, ensure you equally enabled show multiple bonds, too.
1/ Avogadro was able to second guess the bond orders quite reliably
2/ Avogadro2, in ball and sticks with multiple bonds activated, can guess while drawing the structure if it is a single or double bond.
So something seems off.
Note, I use Avogadro for DFT simulations, .mol and .sdf files are not useful in that case, and the additional step of using xyz2mol is just not practical to export graphics of the converged structure and/or make modification to the structure.
I really like the convenience offered by Avogradro.
Avogadro v1 uses Open Babel to deduce bond orders. Avogadro v2 reads XYZ files natively, but this meant that I had to re-implement bond order perception (in 1.97 and 1.98).
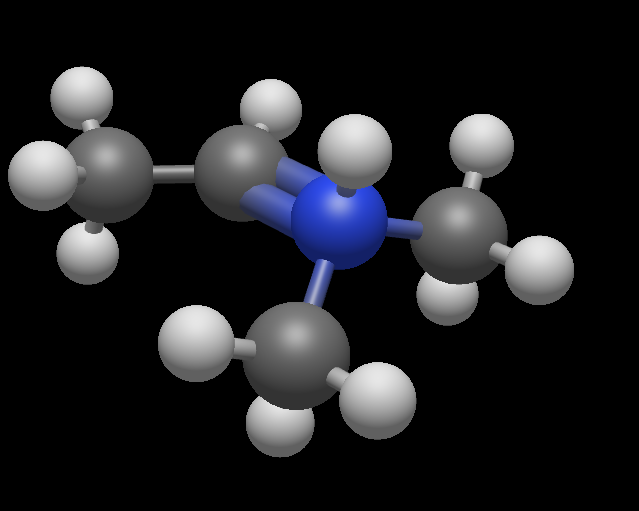
Another observation: the adjust nitrogen behaves oddly.
Description:
Open Avogadro, draw a molecule with just single bonds attached to a nitrogen, e.g. N(CH3)3. Later add one double bond in between N and one C.
The nitrogen then obtain 5 bonds. See attached screenshot
Version tested: 1.95.1 debian package and 1.97 AppImage, Linux Ubuntu 20.22
I mentioned xyz2mol as an example to (re)establish bond orders because reading less than 1k LOC may convey the concept than the larger code basis of Avogadro. That’s it.
If you work in a group which is fine to use .xyz, and the input/output formats of the quantum chemical programs; go for it. In groups set up at the interface of (synthetic) chemistry and physics, I witnessed molecular structures being stored in a group shared database, and by default equally with richer content (at least .sdf and .cdxml) – regardless if the structure lived only briefly in silico, or is a chemical in someones’ shelf (inventory).
Weird. Can you please post that as a bug report on GitHub. I’ll have to check where that falls … Drawing the double bond should promote to N+ (i.e., formal charge +1) … but a quick look shows a formal charge of zero. Seems like possible a few issues in formal charge / valence assignment.