Avogadro version: 1.2.0
Operating system and version: Windows 10 64 bit
Expected Behavior
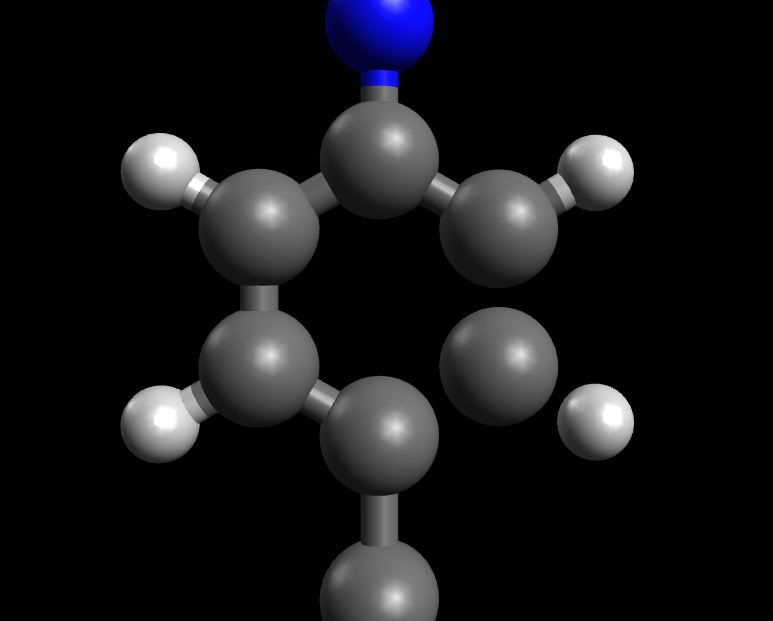
Bonds not detected and displayed from an .xyz file.
I have found this is an increasing problem. The latest example I have an aromatic ring with four of the C-C bonds complete, but the final two are not displayed, despite being the same length as all the rest. The disconnected atom is also disconnected from the H atom it is meant to be bonded to. There are other bonds missing that would also be expected, but aromatic ring is the most important one.
Actual Behavior
Bonds not displayed.
Steps to Reproduce
I am not certain what I need to do, but the file I am using appears to consistently have this problem.
Please upload files if appropriate here (or via file-sharing service like Dropbox or Pastebin) - how to upload files here?
The distances between those particular carbon atoms are ~1.1Å so the bond perception algorithm is very much unhappy. I have no doubt that it tried to assign triple bonds around the ring, plus the hydrogen and couldn’t make it all add up. (A typical CC triple bond would be ~1.2Å.)
The bond perception algorithm isn’t perfect, but if the tolerance is made too loose, a lot of bad PDB / crystal structure files end up with bad bonding too.
Can I ask what program / method produced this file?
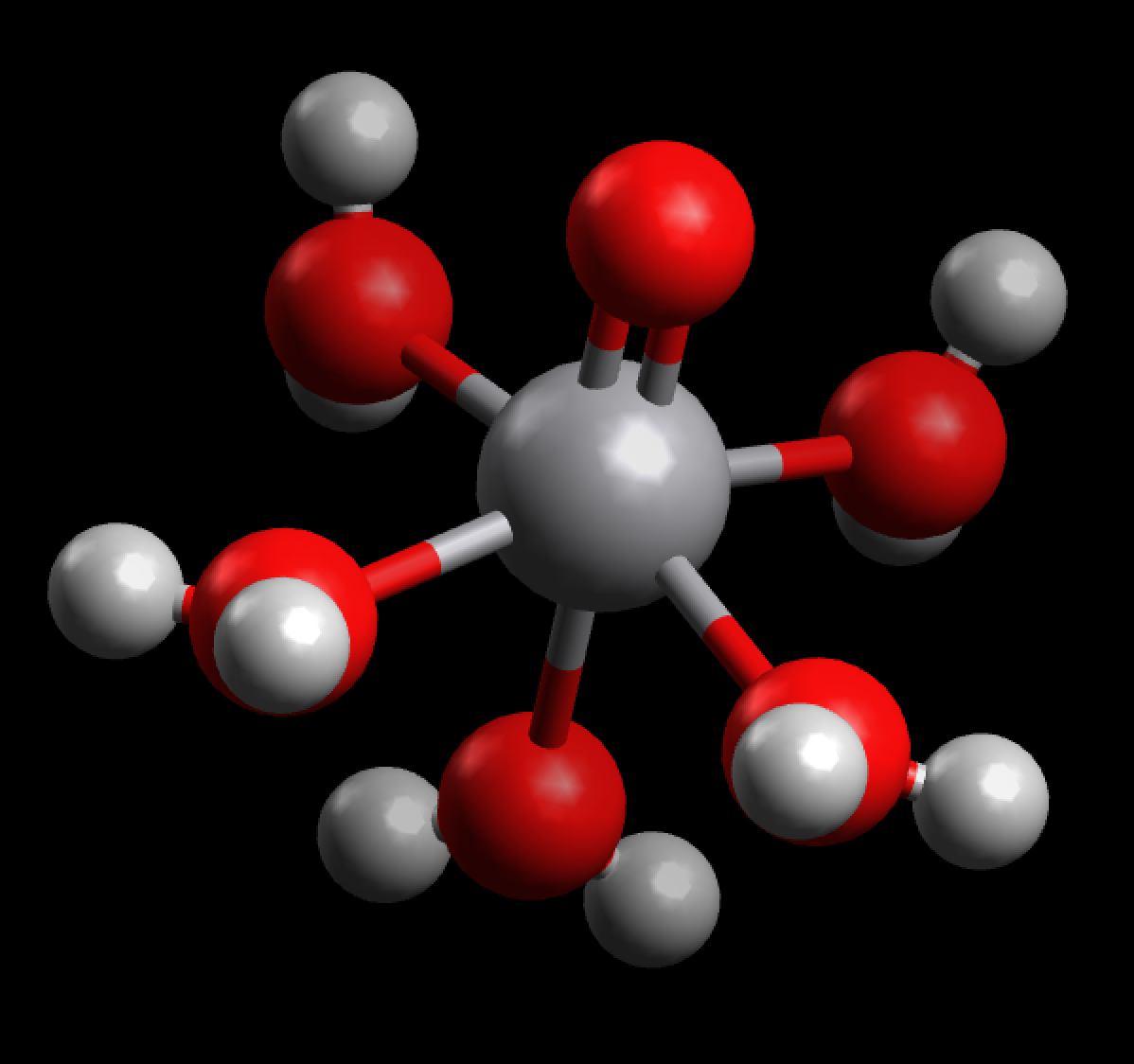
I am having this same problem because I am an inorganic chemist and the molecules I want to display have metal-nonmetal bonds that, I imagine, are longer than the standard C-C et al. bonds set by Avogadro.
Here is a simple example: [VO(H2O)5]2+ xyz file that has 5 V-O bonds that do not show up, but the O-H bonds of course do, as does the V=O bond, which is relatively short.
How can I change the length at which a bond is drawn? How do I set these? I need to add M-X bonds of various sorts and have no idea how.
here is the xyz file, which looks fine in Mercury (all bonds shown):
17
orthogonal from SIBGEQ
V -0.184408 -2.290057 -2.348140
O -0.184408 -2.290057 -0.770933
O -0.184104 -2.509041 -4.512401
H 0.519770 -2.675206 -5.007901
O 1.220904 -0.873215 -2.699066
H 2.096969 -1.015318 -2.851218
H 1.180298 -0.125912 -2.346553
O -1.644841 -3.678667 -2.574867
H -1.896353 -4.123996 -1.862834
H -1.628546 -4.238043 -3.274873
H -0.887512 -2.677165 -5.007901
O -1.593660 -0.877133 -2.699066
H -2.469326 -1.021675 -2.851218
H -1.555135 -0.129720 -2.346553
O 1.279884 -3.674596 -2.574867
H 1.532635 -4.119223 -1.862834
H 1.265147 -4.234014 -3.274873
Avogadro (through Open Babel) uses heuristics about bond detection.
Your case is a bit different… The “problem” isn’t so much the V-O bonds, but rather that the ligands are water. The code detects that H2O fulfills the valence on the oxygens, and won’t add additional bonds to the V atom.
You can manually draw in bonds (make sure the ‘adjust hydrogens’ is unchecked).
I figured that I could manually draw in bonds, but could not see how (in the Build menu, or anywhere else). Where /how is this done? I don’t see “adjust hydrogens” anywhere either.