There is a 2015 post about this but no one answered. I know you can generate a “Xx” point at the center of mass but is there a way to translate the molecule to it?
Relatedly, now I remember there was something strange happening with the calculation of the center of mass…
Defining centers of mass wrt given atoms
I’m pretty sure that’s been fixed.
Use the align tool. Click on the centroid and click align. It will translate everything so that the selected atom is at the origin:
If you only click on one atom, the align tool translates that to the origin. If you click on two atoms, it will align the second atom with the specified axis.
My 1.102.1 still places the CM outside of the internuclear axis for the NO example I was using before.
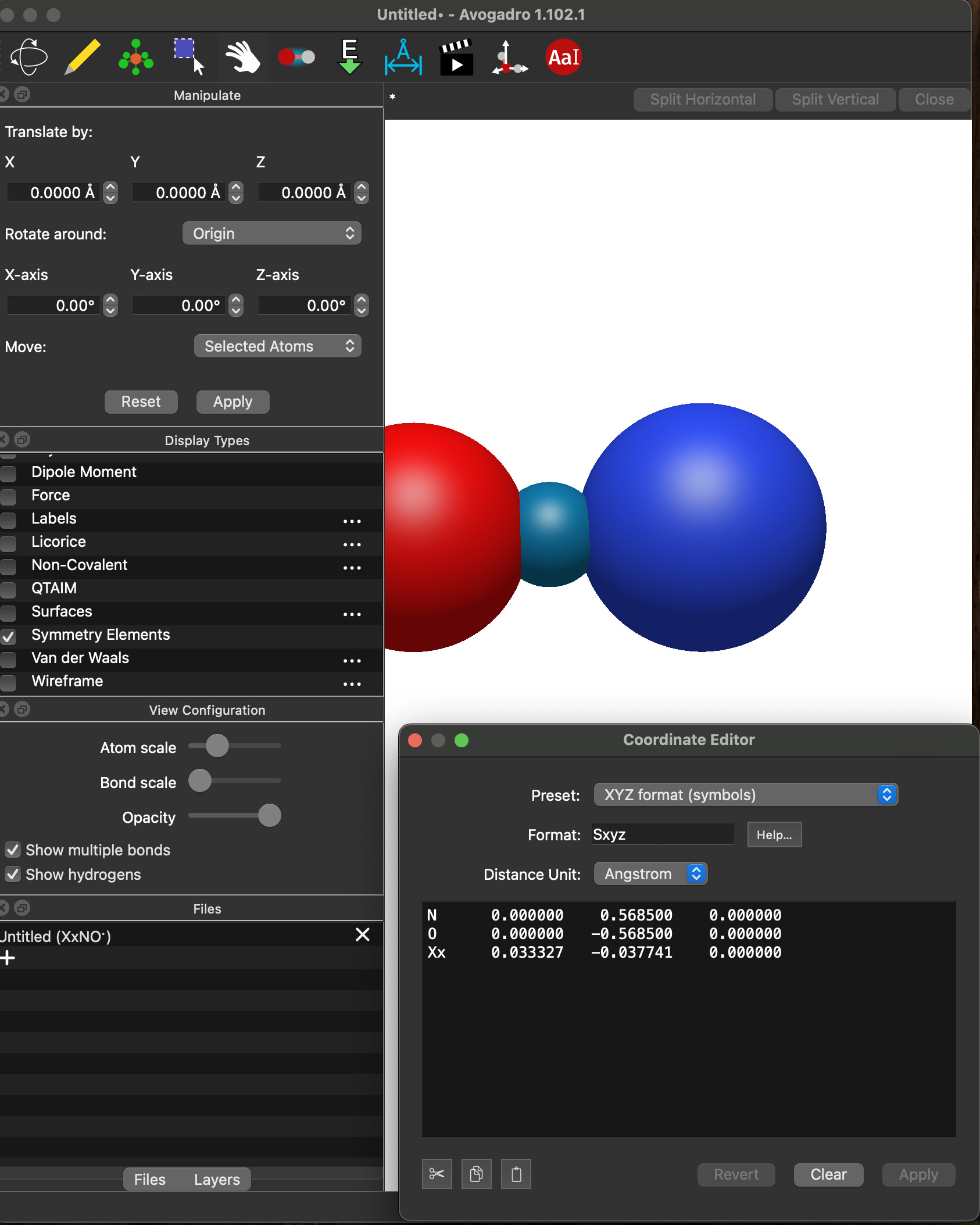
I’m scratching my head. I can’t reproduce that at all.
I just entered your exact system, (i.e., the N and O atoms at those positions) clicked “Add Center of Mass” and got
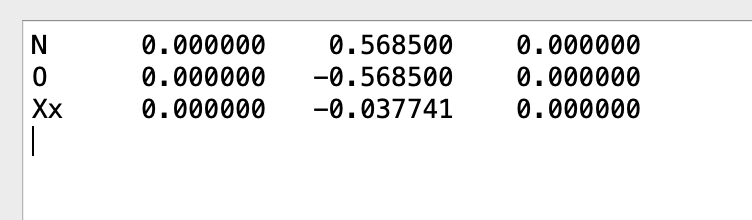
I’m also looking at the code, and I don’t see any way you’d get an x-component:
I’ve been trying a few orientations and I can’t get that to happen.
Can you send me the NO example without the center-of-mass added so I can check it? Maybe there’s something I’m missing.
You mean an output file? I have none - I build the molecule from the Cartesian editor. So something interesting happen - sometimes it works correctly and some time it doest not. Here is a .gif of when it worked.
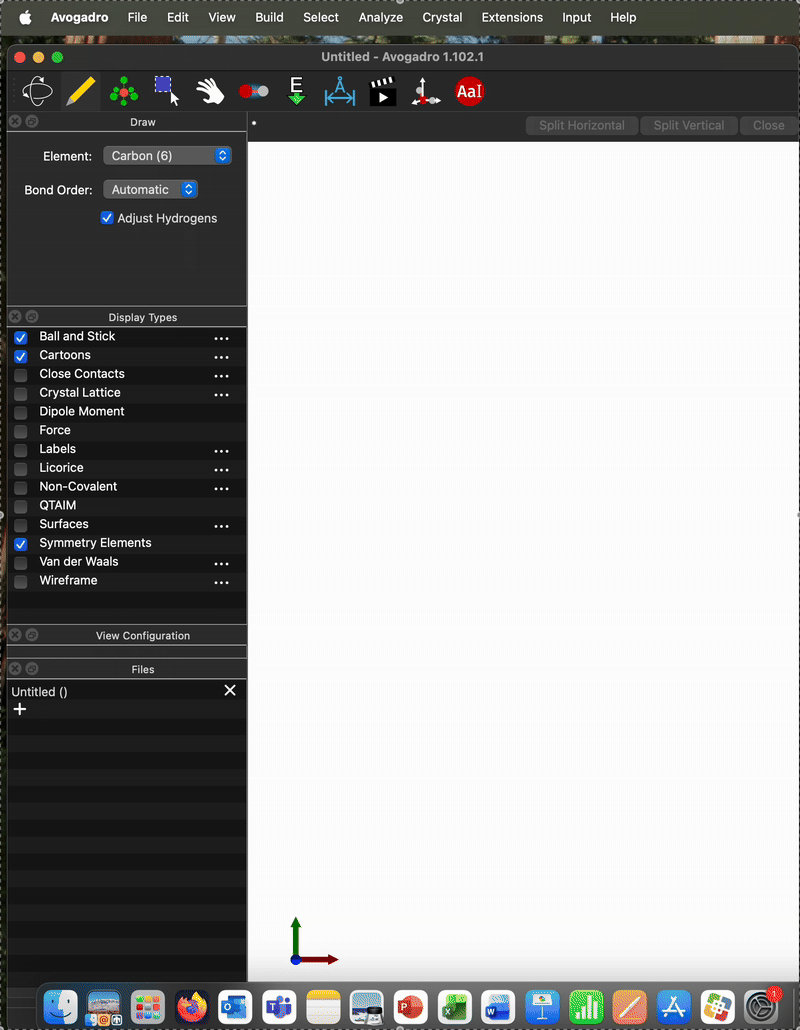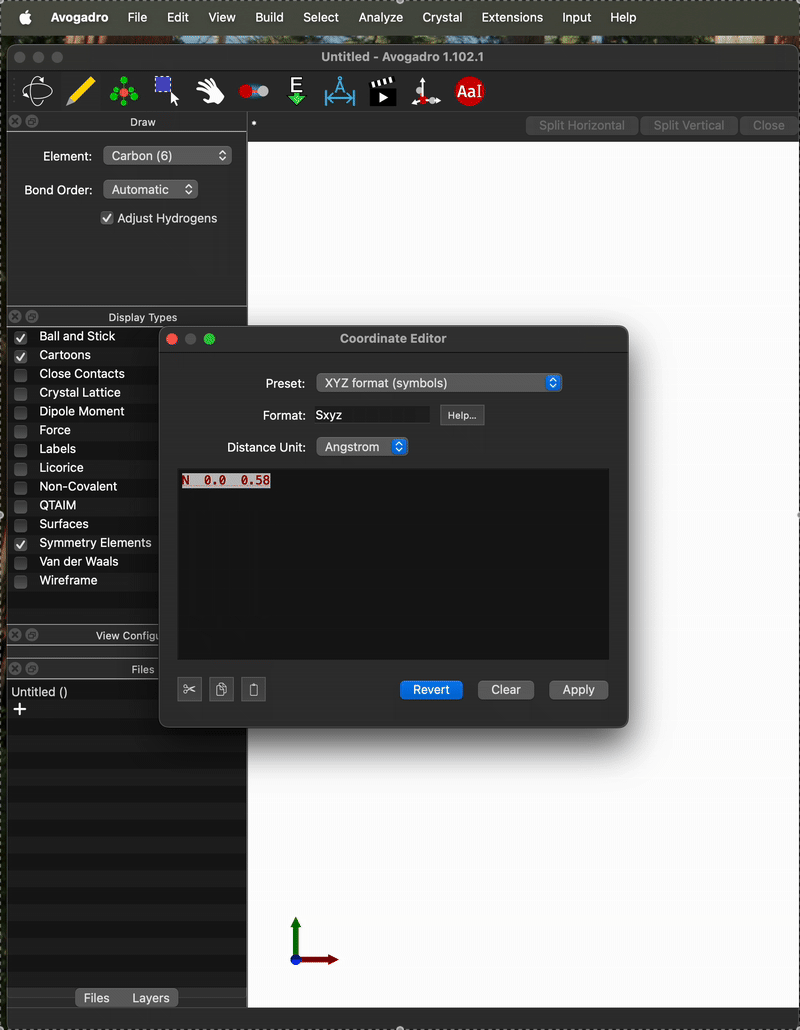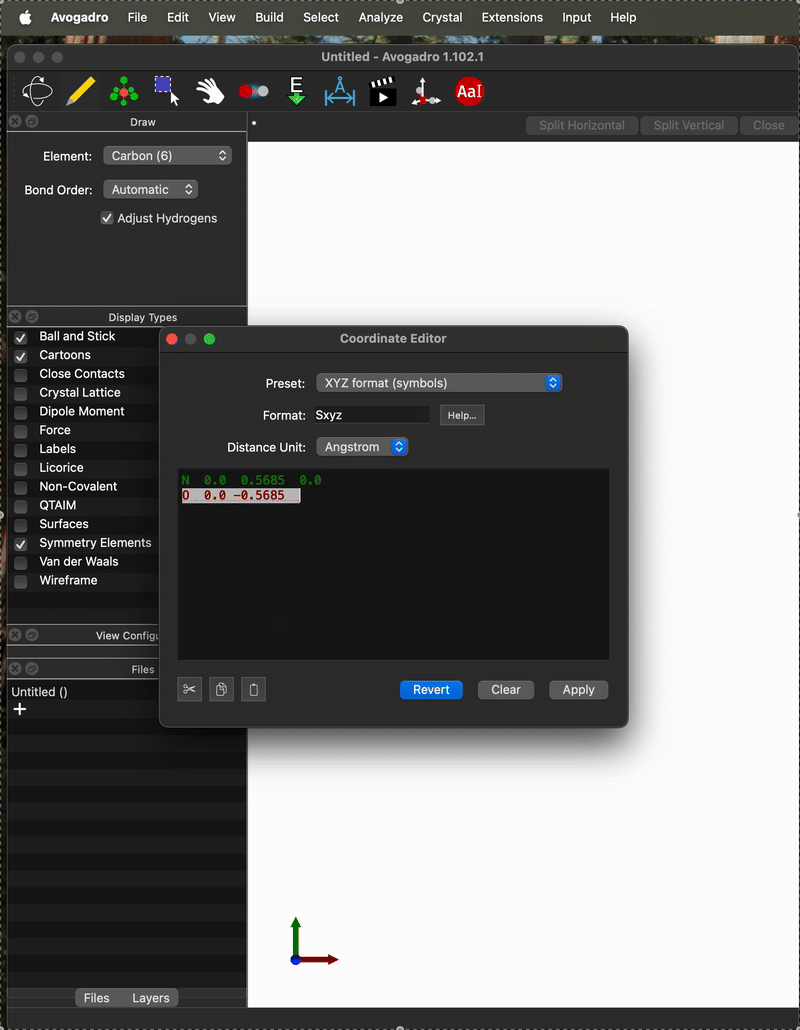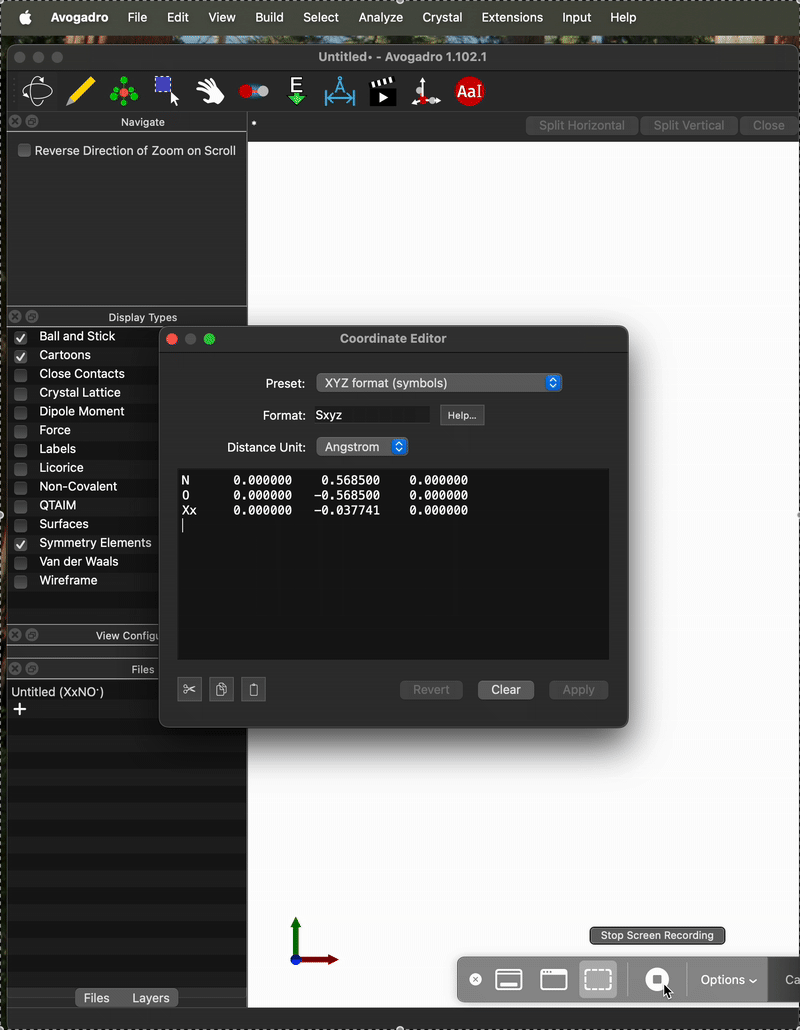
And here is an example of when it is not. One difference I can see is that on the “working case” I entered the coordinates as:
N 0.0 0.5685 0.0
O 0.0 -0.5685 0.0
but on the not-working case I entered them as
N 0.0 0.5685 0.0
O 0.0 -0.5685 0.0
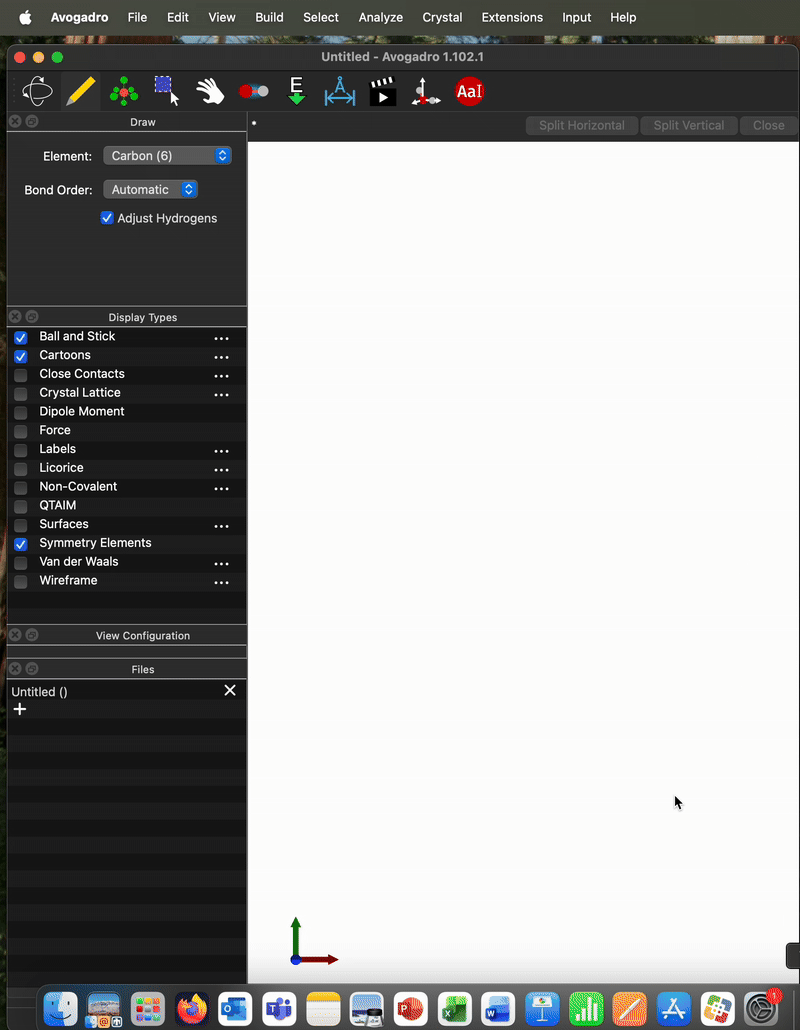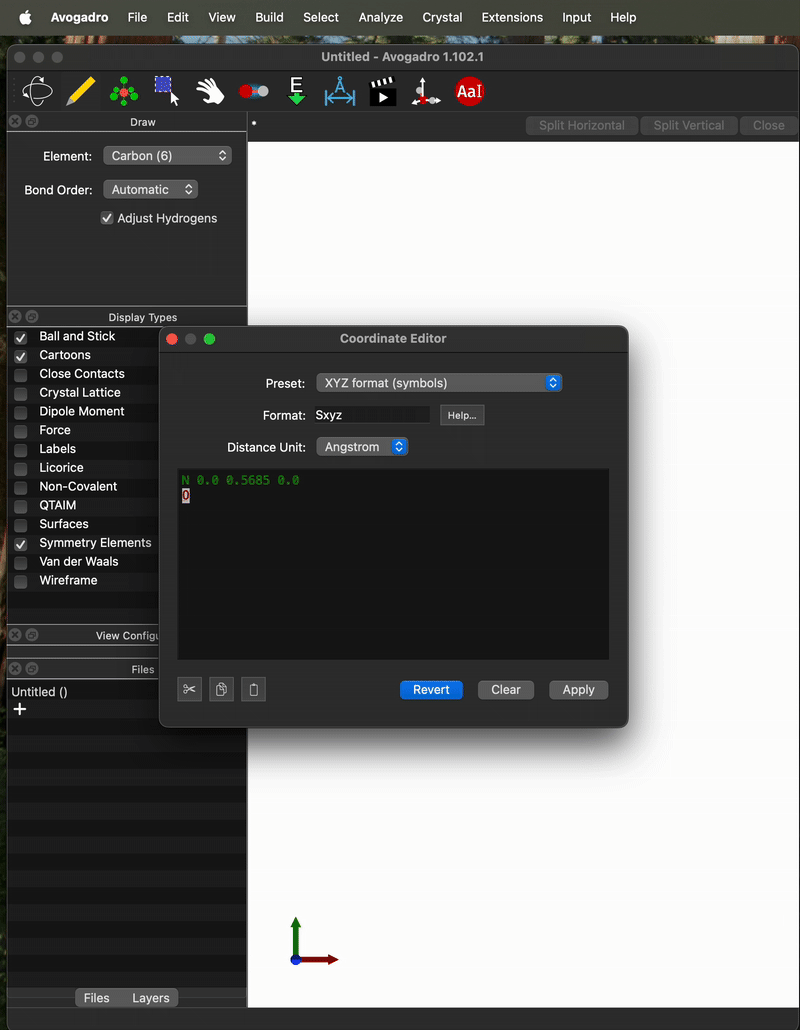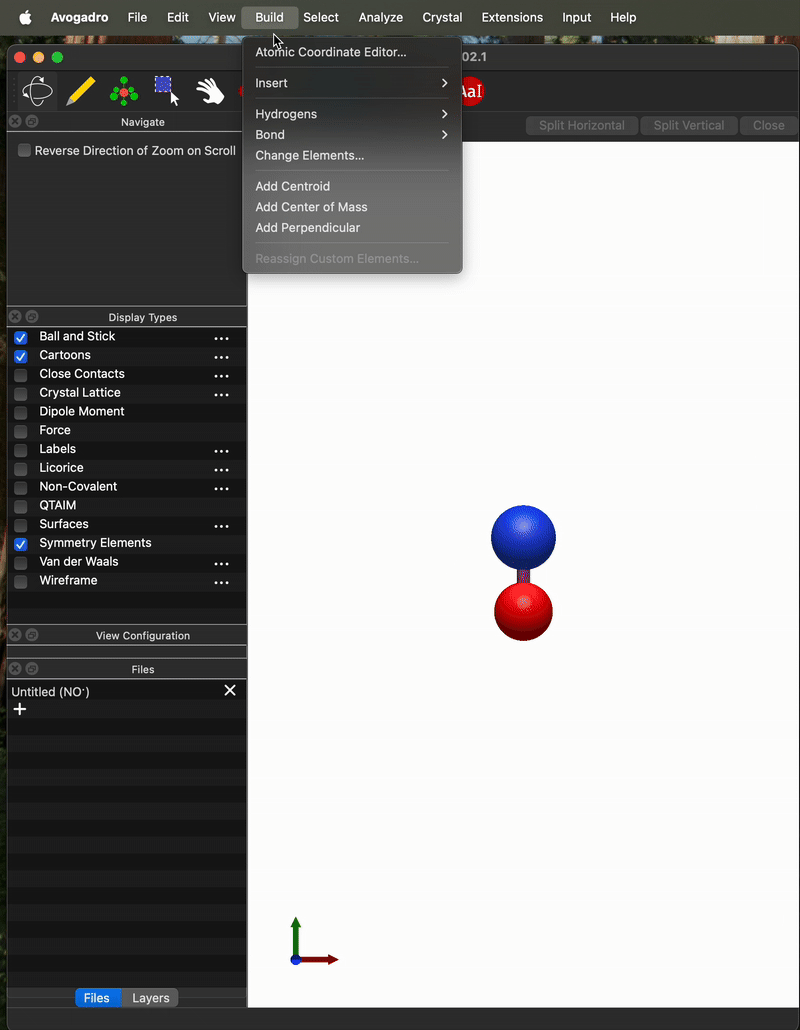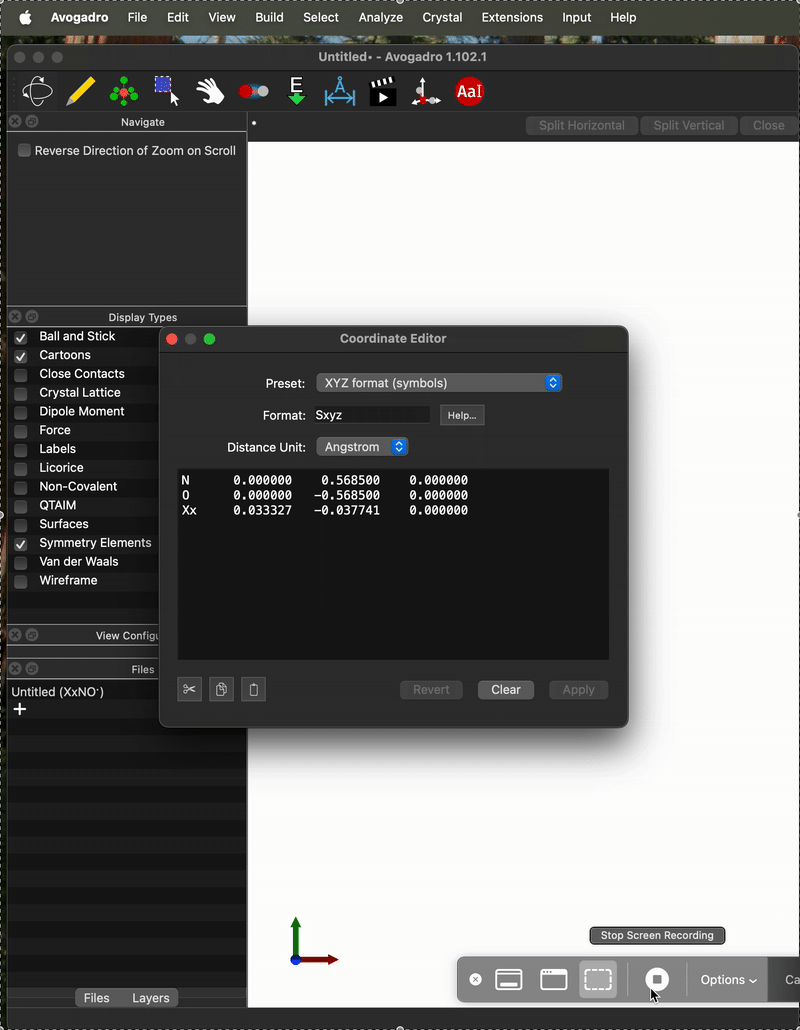
Here’s an .xyz file for a moleculer that I manually translated to the CoM. When I open it and calculate the CoM with Avogadro I get 0 on the y and z coordinate but I get the same 0.003332 as NO) on the x coordinate.
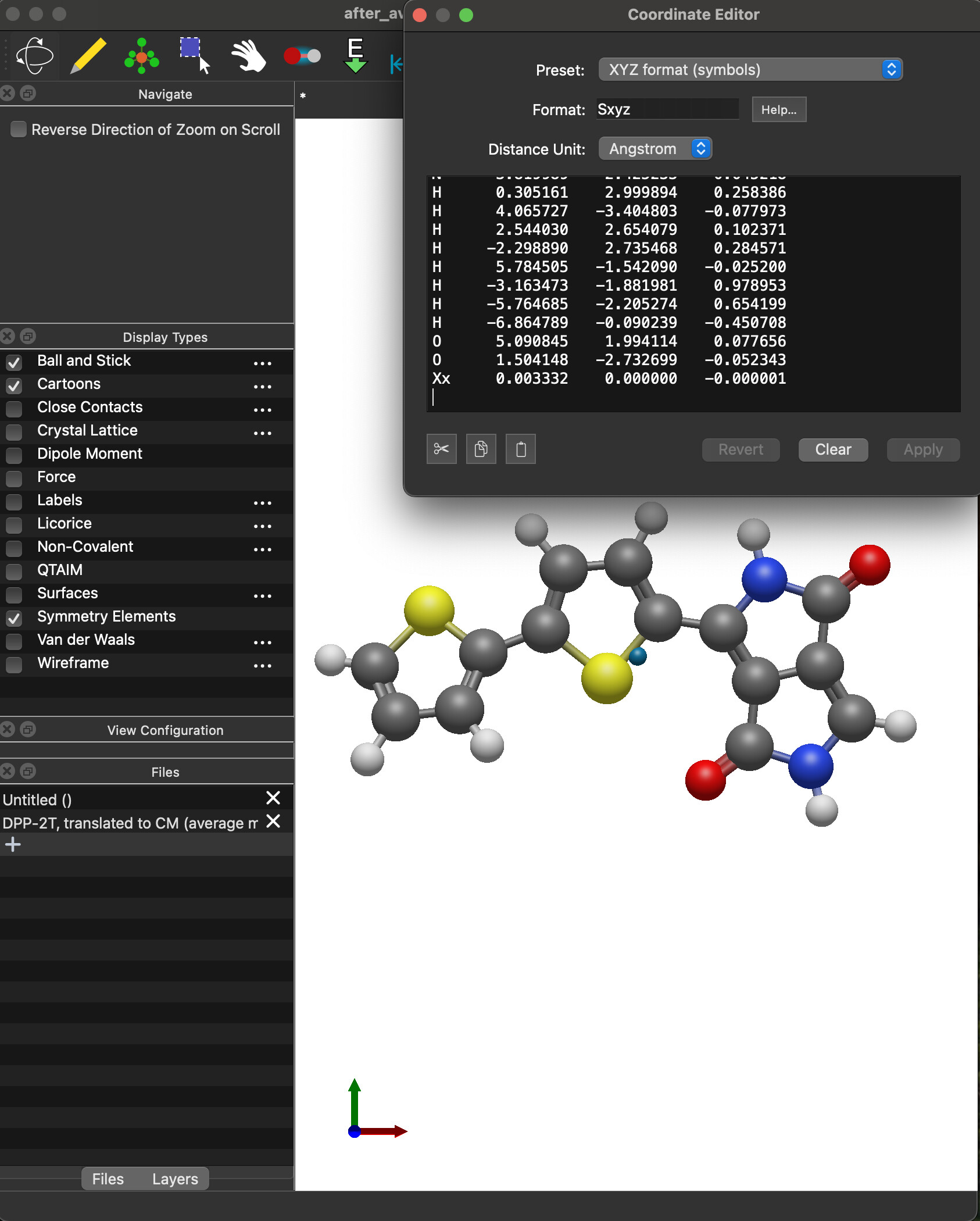
after_average.xyz (1.1 KB)
This feels like some sort of weird compiler optimization bug (e.g., numeric noise). For example, I absolutely cannot reproduce it on my M4 MacBook, but my M1 desktop shows it sometimes but not all the time.
As far as the underlying code, there’s no difference between the two examples you gave, or N as first atom and O as second or vice-versa.
Yikes… I suppose for now I’ll not use this functionality. It’s easy to see for NO but for more complex systems I wouldn’t be able to tell if the CoM calculated by Avogadro is “a little off”. This is relevant to me because I’m working on a project involving external electric fields and I need my molecules placed and oriented in very specific ways.
Give me a bit - I’m putting in a new Mac build that at least in the times I’ve checked it doesn’t have problems.
As you might imagine, we do plenty of calculations in electric fields, although we often script the preparation in Python.
It’s one reason for the align tool.
1 Like
Okay, give this Mac ARM build a try, or wait ~20 minutes for the continuous build to update:
https://github.com/OpenChemistry/avogadrolibs/releases/download/continuous/Avogadro2-continuous-arm64.dmg
1 Like
Ok I think it works! That same .xyz file does not show the strange 0.003332 for the CoM in the x coordinate.
One question that remains for me is: what is the argument to use the average isotope masses for the CoM calculation vs. those of the most common isotope?
At the moment, we don’t have isotopes so it’s going off the average isotope masses. Most common isotopes / specific isotopes will go in soon. For that, there will probably be a command to assign isotopes with the default being the most common.
1 Like