I’m beginning a project that requires me to scan PESs in Jacobi coordinate systems. It would be nice to have some tools to visualize these in Avogadro.
I envision something like being able to visualize the center of mass with respect to some given atoms, and then being able to define distances and angles with respect to it. In this simple example, I would like to see the center of mass of the NO diatom and then measure the distance to the Cl from it, as well as the angle set by the CM-N and the CM-Cl vectors.
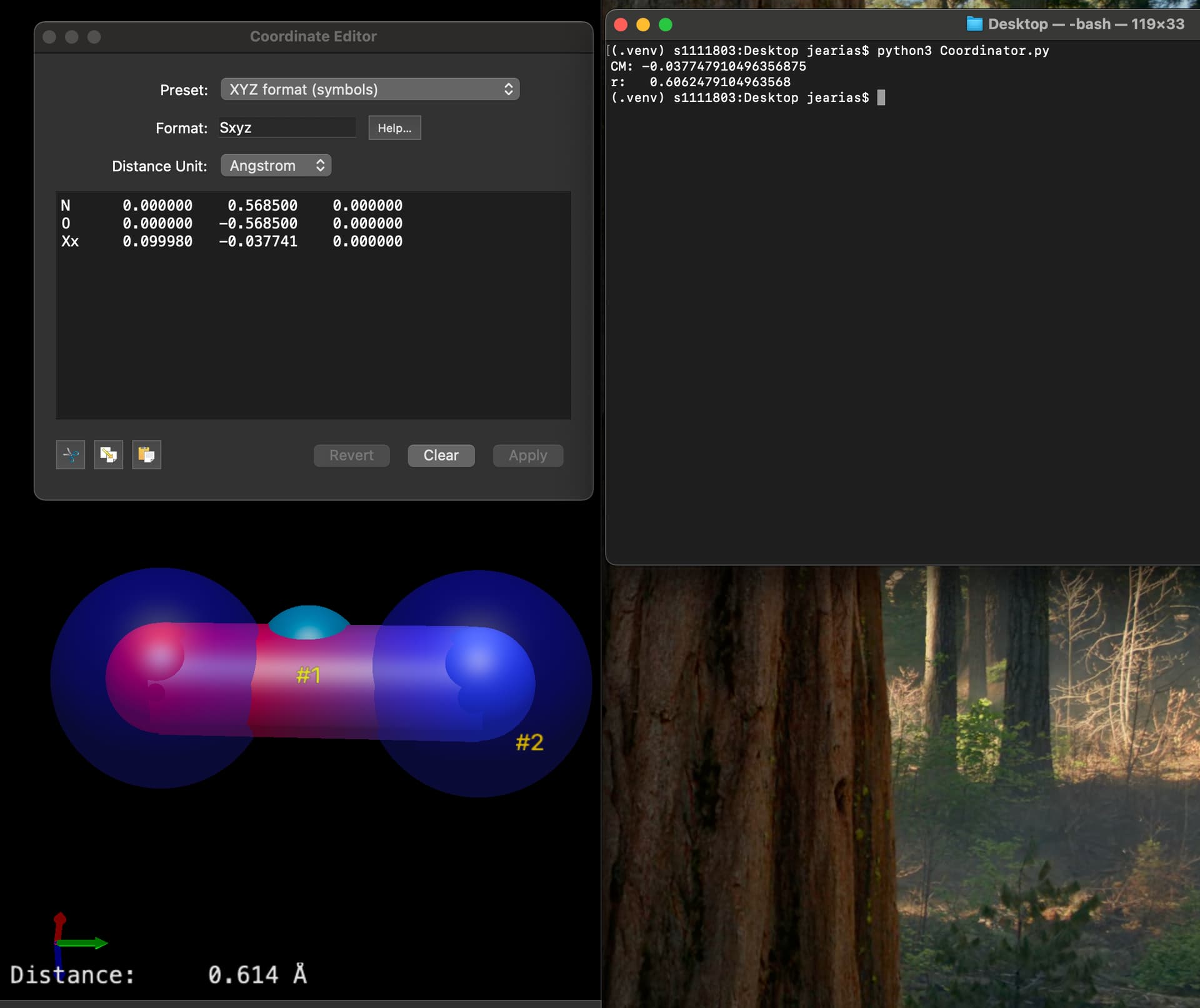
When I build N and O on the cartesian editor along the y axis, equi-distant from the origin, and generate the center of mass, it places it outside the y axis. Naturally, distance from the CM to any of the atoms is not what I would calculate by hand.