I think this might be an issue with Avogadro, although it might be a user error. There might be a plugin or something I need to install.
Environment Information
Avogadro version: 1.96.0 (QT 5.15.4)
Operating system and version: Ubuntu 20.04.4
Kernel: 5.4.0-109-generic
Expected Behavior
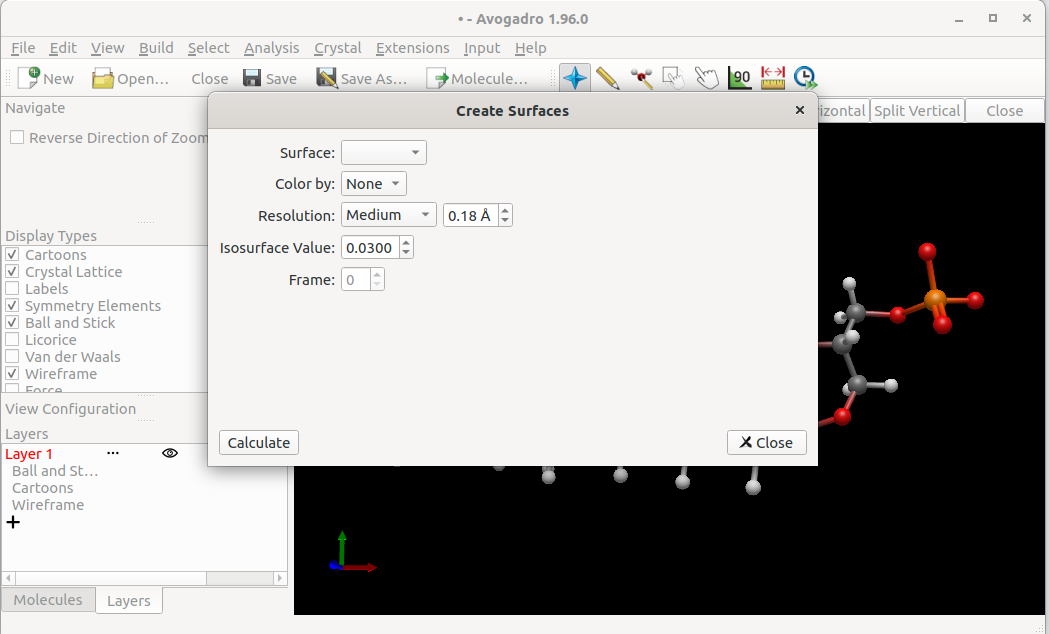
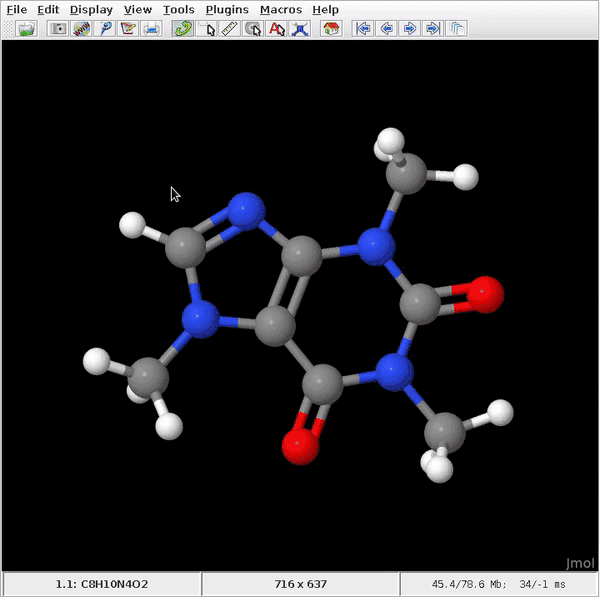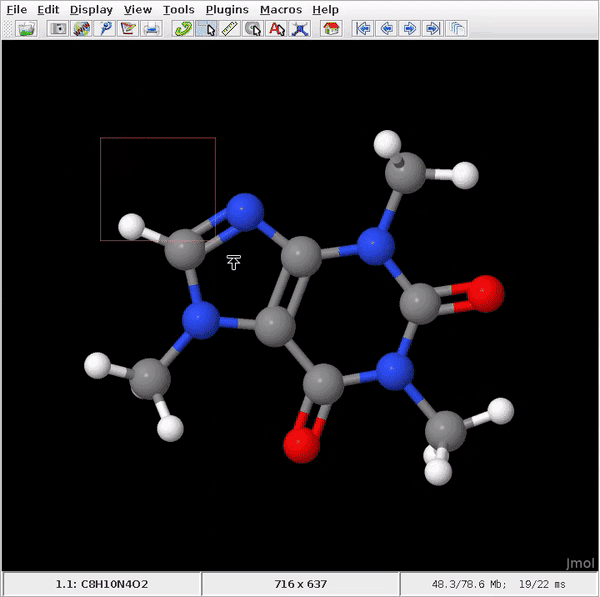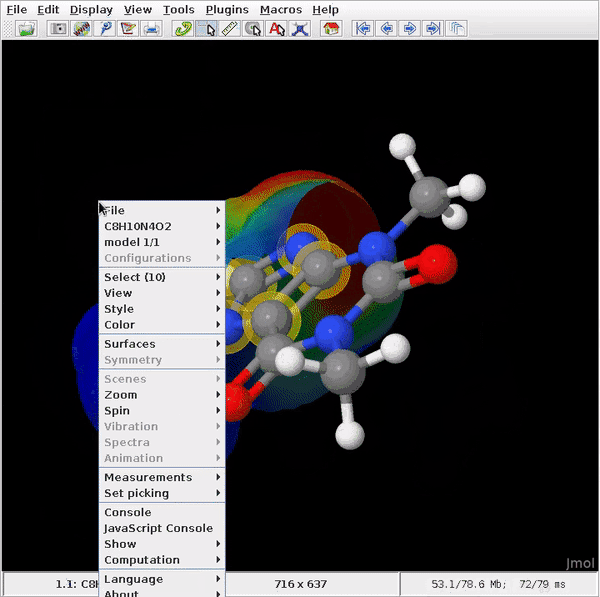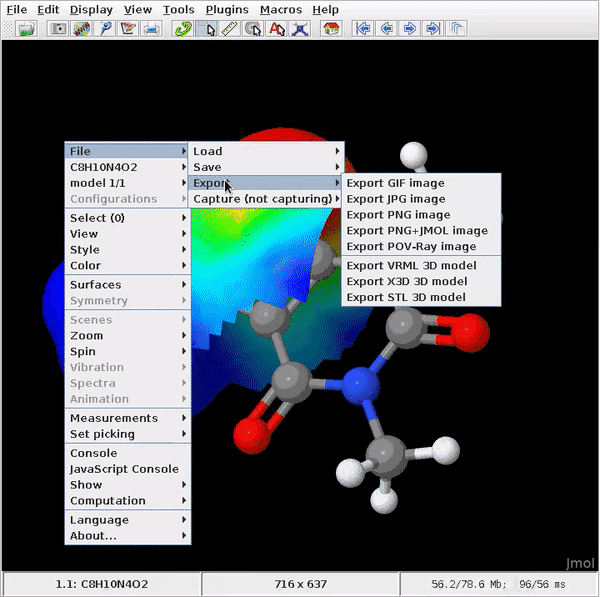
After creating a molecule, going to Analysis > Create Surfaces…, I should be presented with a dialog with a menu for surface types with different surface types (such as Van der Waals, for instance).
Actual Behavior
Unfortunately, while the dialog does show up, the drop down is non-responsive.
Affirmative, the same observation (void selection of surface) when running the Avogadro2-x86_64.AppImage for Linux 1.96.0 (mentioned here, download per 2022-06-04 a few days earlier) in Linux Debian 12/bookworm (branch testing).
I appreciate the “replication study” as it were, @Thomas. You wouldn’t happen to know if there is anywhere I could be looking for more information on what might be causing this? Log messages, code in a certain module, etc. I’m still pretty new to Avogadro.
Running Linux Debian 12/bookworm (branch testing), I just run the installation of Avogadro 1.96.0 as provided from the OS repositories via synaptic; same result (analysis → create surfaces is void).
When presenting the current AppImages (reference), Geoff pointed to a public GitHub repository. If you have command of C++ (right hand side, section languages), perhaps you spot the road block.
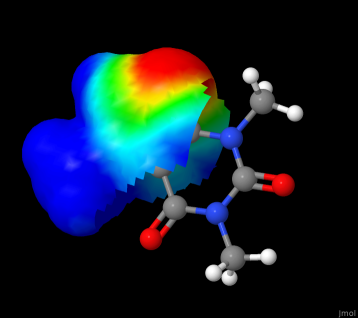
In case your aim is a quick computation of van der Waals surface, or to display molecular electrostatic potential (which is my current goal), let the maintainers have all the time they need; for now, I resort to Jmol as an alternative/bypass.
The .gif below provides some impression (but in post production was reduced to 64 colours and low resolution to pass through the size limit in this forum, 4MB per file.)
AH I see. I’ve plenty of experience making bad code, not on purpose, so I sympathize. That said, it also must be pretty frustrating as a team working on an open source project. I used to work as a developer, but as you might guess I was not particularly good at it in my estimation, and in any case I did not particularly enjoy it. The experience does give me a good understanding of how difficult the process can be.
I really love that this exists and would love to help, if I can. It’s a little intimidating as someone still learning chemistry, I have to say. I’ve found some tutorials online and am trying things out. Perhaps I can help with some of the user documentation at some point.
Bug reports, feature suggestions, documentation …there are always lots of ways to help in a community project.
I’ll be posting more about help needed for user documentation later this summer. (Once a few of these missing pieces settle into place… always difficult to take screenshots when the UI changes a few weeks later.)