I tried doing this but am not understanding whether its correct
Code:
def Rotate(opts,mol):
angle=float(opts['angle'])
atomic_numbers = mol['atoms']['elements']['number']
selected_atoms = []
selected_coordinates=[]
atom_names=[]
for i in range(len(atomic_numbers)):
if mol['atoms']['selected'][i]:
selected_atoms.append(i)
atom_names.append(name.get(atomic_numbers[i]))
coords = mol['atoms']['coords']['3d']
j=0
for i in range(0, len(coords), 3):
if j in selected_atoms:
selected_coordinates.append(coords[i])
selected_coordinates.append(coords[i+1])
selected_coordinates.append(coords[i+2])
j = j+1
operation_list = OperationList()
coordinates_array = np.array(selected_coordinates).reshape(-1, 3)
selected_geometry = GeometryXYZ(
names=atom_names,
coordinates=coordinates_array,
comment="Best-Fit Selection"
)
selected=[]
for k in range(len(selected_atoms)):
selected.append(k)
translate_to_origin = CentroidTranslate(selected, fac=-1.0)
operation_list.append(translate_to_origin)
rotate_to_xy_plane = NormalRotate(selected,angle)
operation_list.append(rotate_to_xy_plane)
rotate = ShiftedOperation(translate_to_origin, rotate_to_xy_plane)
operation_list.append(rotate)
for operation in operation_list:
operation(selected_geometry.coordinates)
for i, idx in enumerate(selected_atoms):
start_idx = idx * 3
end_idx = start_idx + 3
mol['atoms']['coords']['3d'][start_idx:end_idx] = selected_geometry.coordinates[i]
return mol
echo '{"angle":30,
>> "cjson": {"chemicalJson": 1,
>> "atoms": {"coords": {"3d": [0.617532, -0.027246, 0.481009, 0.156663, 0.031752, -0.362419, -0.774185, -0.004516, -0.11859,0.617532, -0.027246, 0.481009]}, "selected":[1,1,0,1],
>> "elements": {"number": [1, 8, 1,4]}},
>> "bonds": {"connections": {"index": [1, 2, 1, 0]},
>> "order": [1, 1]}}
>> }' | python rotation_script.py --run-command
Output:
{"cjson": {"chemicalJson": 1, "atoms": {"coords": {"3d": [0.11930055667962669, -0.015581411770221364, 0.29757784441826723, -0.23860111335925333, 0.031162823540442783, -0.5951556888365344, -0.774185, -0.004516, -0.11859, 0.11930055667962669, -0.015581411770221364, 0.29757784441826723]}, "selected": [1, 1, 0, 1], "elements": {"number": [1, 8, 1, 4]}}, "bonds": {"connections": {"index": [1, 2, 1, 0]},
"order": [1, 1]}}}
So I have applied Rotation only to the selected atoms.
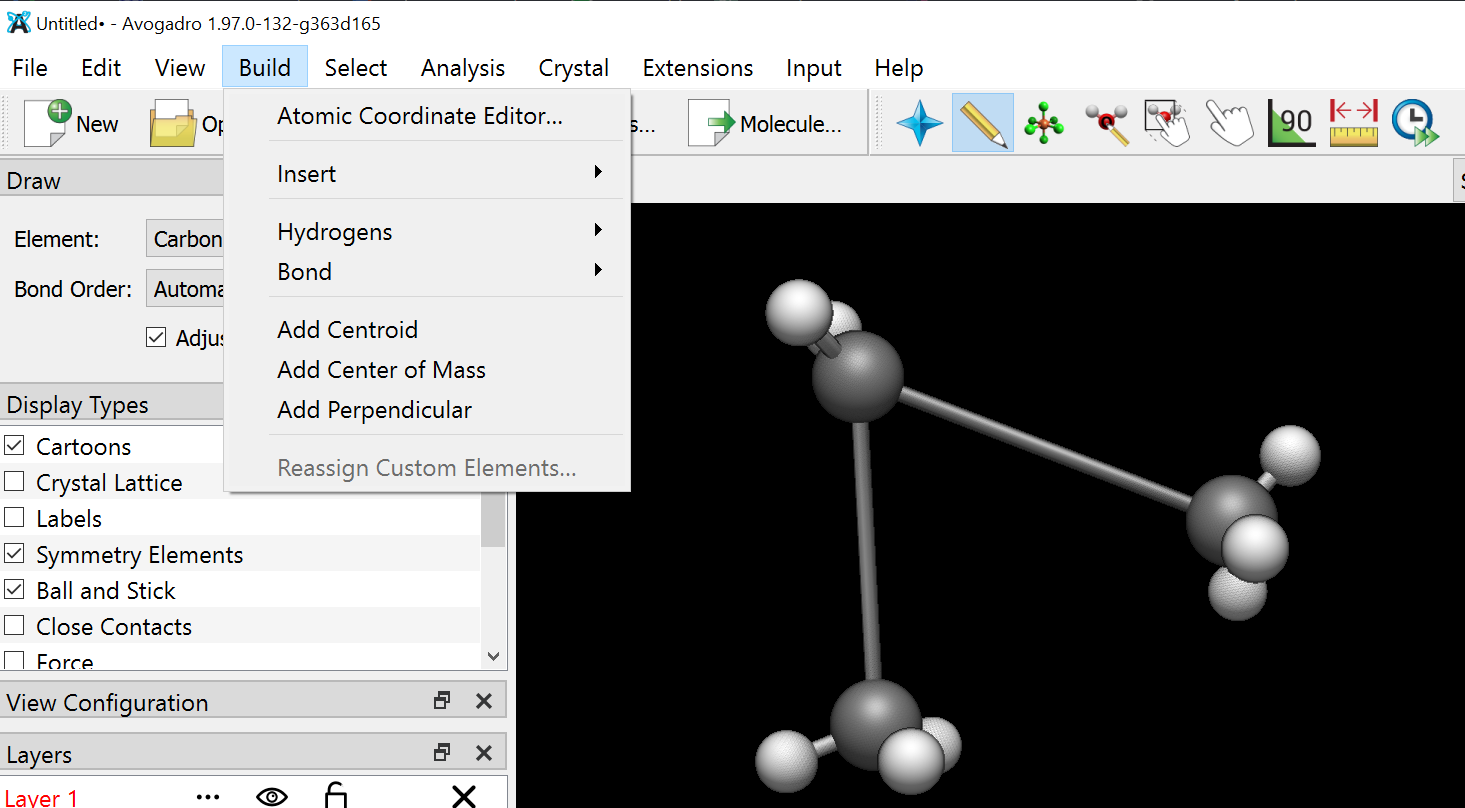
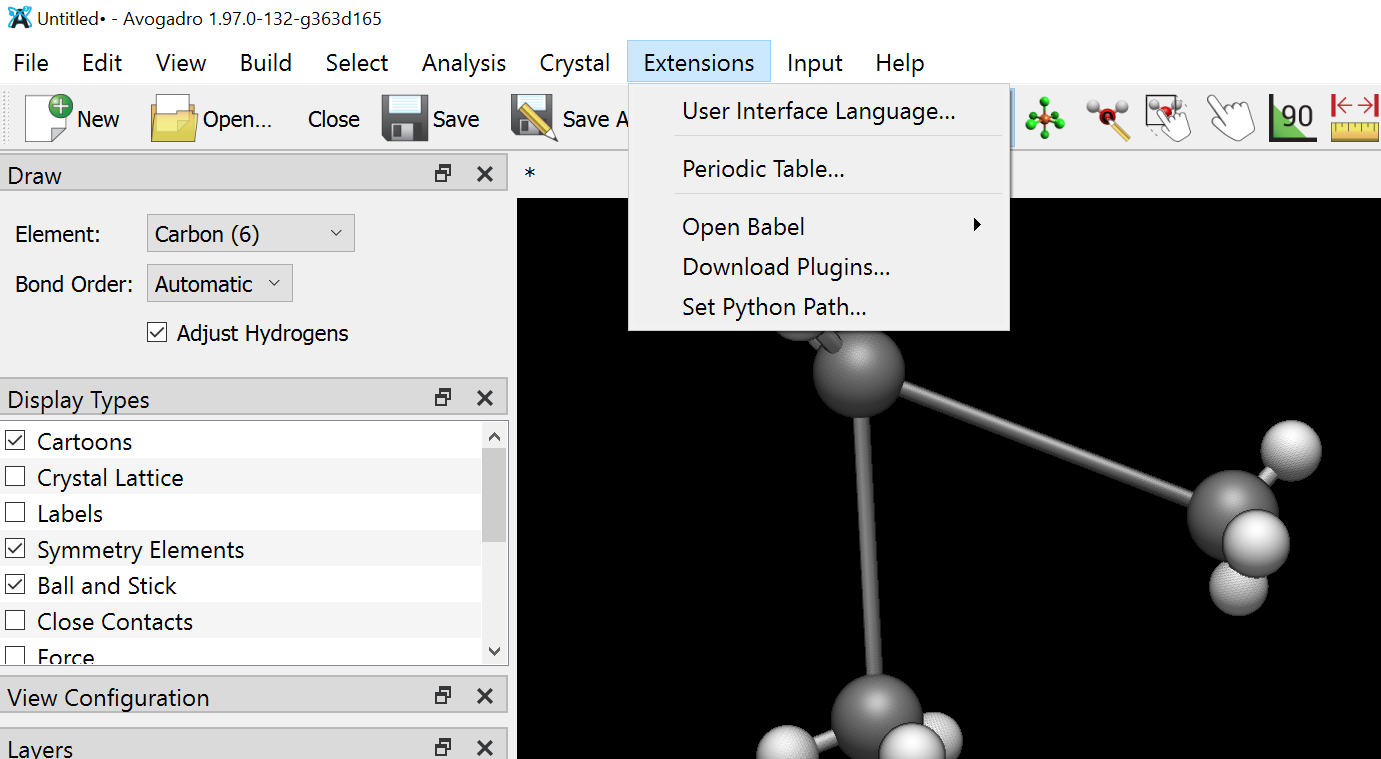
Also can you please tell me how can I visualise the script on avogadro because when I try to drag and drop…The script gets installed but I dont know from where can I access it
Thank You