@Juanes Your first post remains vague what your eventual intent of (now I speculate:) quantum chemical calculation is .and. what program you then want to use.
One of the problems «other» non-crystallography programs face when managing the information stored in a .cif is that they define i) a structure motif (at the lowest, irreducible level) which ii) by symmetry operators of the unit cell (keyword space groups, of which there are 230 conventional in the International Tables, some only with standard, other with additional non-conventional setting, for instance P2_1/c vs P2_1/a) «complete» the content of the unit cell. Note, the coordinate system in this parallelepiped can, yet need not be one of orthogonal axes (e.g., a cubic vs monoclinic Bravais lattice), nor that the unit vectors in the three directions need to be of equal length (e.g., cubic vs tetragonal Bravais lattice) while e.g., .mol and .xyz (implicitly?) use a Cartesian coordinate system, too. And iii), .cif use fractional coordinates along \vec{a}, \vec{b}, \vec{c} typically in the range of [0.0 \ldots 1.0] because an atom at [1.5,0,0] is equivalent to to one at [-0.5,0.0,0.0] or [0.5,0.0,0.0] but when establishing a crystallographic model it can be useful to use e.g., negative components of one set of atomic coordinates in favour to manage the whole crystallographic motif. And then, 1a is e.g., 3.458 A in one particular model, but 5.88 A in another.
Thus, reading a .cif by a non-crystallographic software can be misleading. Thus e.g., one TCNQ molecule (your second post, left hand illustration) appears «still incomplete». Equally note: a .cif file may, but _need not_define bonds, types of bonds, or bond order as you would find e.g., in the bond block of a .sdf file. If authors do so (e.g., small molecule crystallography/chemical crystallography), they do with a particular syntax managed by the IUCr, for instance with _chemical_conn_bond_type (record)
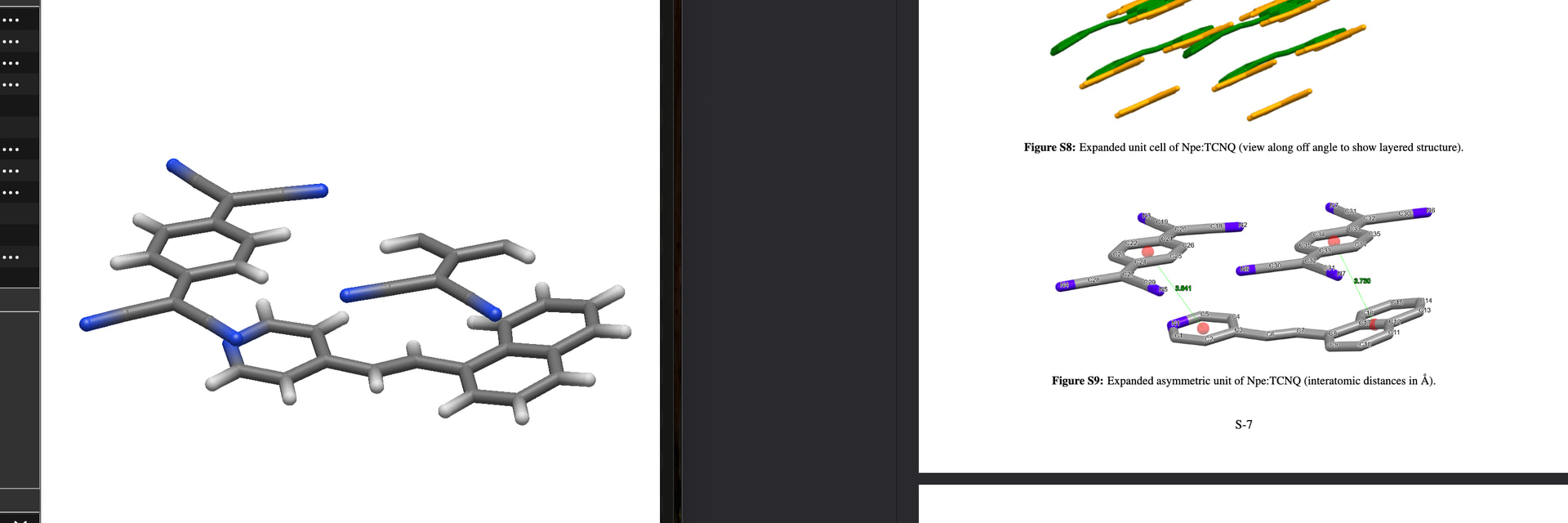
Side note about the illustration S9: the magenta colored spheres are centroids. Depending on the software at disposition, you may select a couple of real atoms, the program then computes the geometric centre of them. Centroids can facilitate the description of intermolecular distances; for interactions (let’s say \pi - \pi between adjacent arenes) you have to include at least relative orientation/angles, too.
What do you want to compute later on? Which program in mind? If Avogadro2 (and openbabel) struggle, use CCDC Mercury (there is a free community edition) for «the completion of molecules» and packing, Jmol with its interface to Gaussian (as an entry, see the tutorial in JApplChem by Hanson, doi:10.1107/S0021889810030256), or Vesta to mention a few GUIs out there. Presuming your are comfortable with the CLI, cod-tools by the COD (and repackaged by DebianChem) provide many helpful utilities like codcif2sdf, cif_fillcell etc., too.