since the release of Avogadro 2 is imminent, I would like to quickly mention a few things regarding the crystallography functionalities.
I think most users would expect that when loading a CIF, the entire unit cell content would be displayed, not just the asymmetric unit as is currently the case.
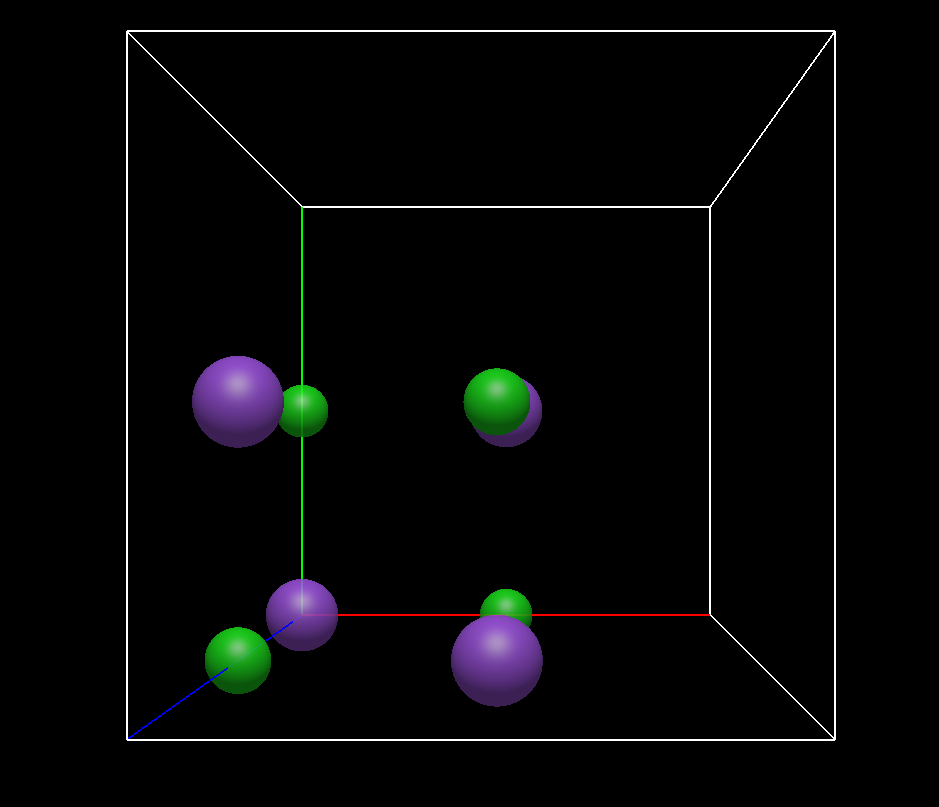
It is unclear to me what the “Wrap Atoms to Unit Cell” command means – perhaps that only atoms with coordinates between 0.001 and 0.999 are displayed? In any case, this command does not work either; only a few bonds are drawn (see image below).
You can find ample discussion of this on the forum (e.g. from @Thomas in particular) . I welcome suggestions.. there are a few requests on the GitHub issues as well.
The problem is that some CIF are molecules (i.e., give me just the asymmetric unit) and some are solids, in which case people want the primitive cell.
I welcome heuristics to differentiate these use cases. Using the asymmetric unit, with the ability to generate the primitive cell easily is the current default.
If you have a good suggestion for “this CIF should show the primitive cell instead of the asymmetric unit” I’d be happy to implement it (e.g. how to detect a molecule vs. a solid)
I’m not sure what you want it to do. That’s the primitive unit cell. Do you want it to include the redundant edge and corner atoms?
The catch of course with that is exporting to other formats (e.g., calculations) in which primitive is needed and the edge and corner atoms are duplicates.
We can certainly add some command like “Fill Edges and Corners” or something.. Suggestions for wording would be helpful.
It takes any atoms outside of [0, 1] and wraps them into the base unit cell. Perhaps that’s a documentation thing, but I’m not sure why you think that isn’t working?
Please don’t take this as critical - I definitely want to see improvements in how we handle crystal structures, solids and materials.
We’ve talked about the heuristics maybe a few times. At the moment, we don’t do our own CIF parsing, so we don’t have easy access to the _chemical_formula_moiety etc. (and sometimes those are missing anyway).
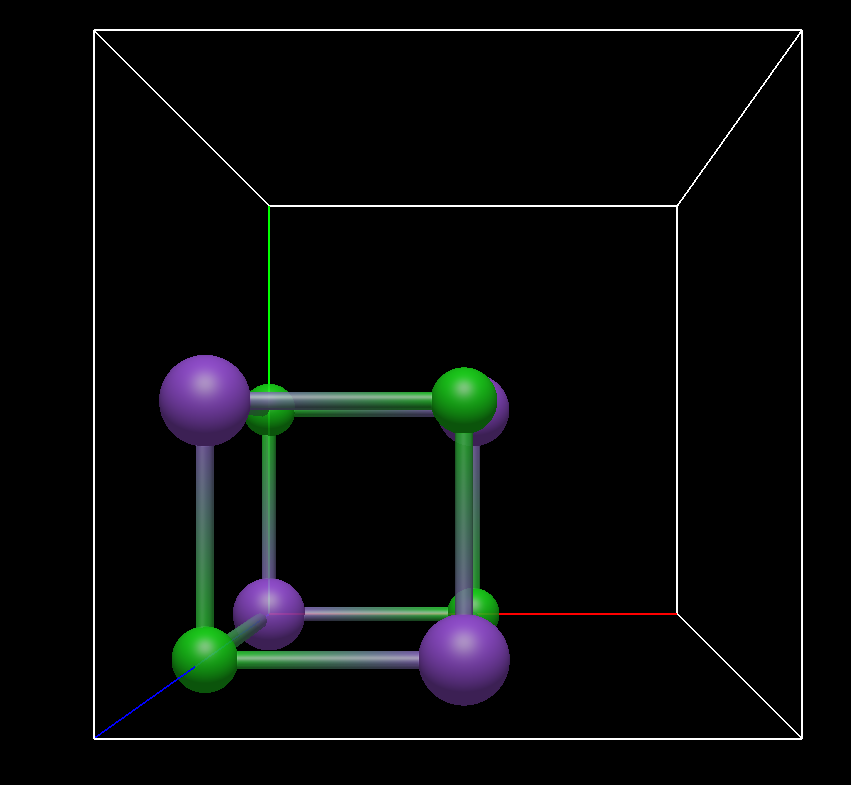
Yes, exactly. That is the common way to display a ‘filled unit cell’. I don’t know of any software designed for crystal visualisation that would do it differently. Take CrystalMaker, Diamond 5, VESTA, Mercury, Materials Studio etc. The way Avogadro it handles is to fill a NaCl unit cell (depends of course on the space group) by 12.5 %, the remaining volume ist empty.
If you look up pictures of unit cells in encyclopedias, crystallography text books, journals etc. usually the unit cell is actually really (completely) filled. If the cell is centered, like in the case of NaCl, I can imagine some purposes for which you want so see the primtive cell, but I would rather go the other way round and would fill the whole unit cell as default (including redundant atoms) and include a command ‘Show Primitive cell’ and ‘Show Asymmetric Unit Only’ or something like that.
In my humble opinion most of the users (crystallographers ) have a intuitive understanding that filling means actually filling completely. My suggestion is described in my last paragraph:
Fill the Unit Cell/Display Complete Unit Cell
Show Primtive Cell
Show Asymmetric Unit only
Ok, I couldn’t have anticipated that because in the NaCl case, there was nothing outside the cell, so that nothing happened, except that bonds were generated (why is this the case?)
Bond generation would be another topic
Other topics are also some of the unit cell commands, but I will write some thoughts about these in the next few days.
We had this discussion not too long ago. The problem in some sense is the overlap in use cases. Yes, displaying unit cells typically shows all the edges. But someone who saves to VASP, etc. only wants the primitive conventional cell.
I forgot something. Maybe it’s not important, but if you write that the primitive cell view was implemented to transfer it to quantum chemistry applications, then maybe it is important after all:
From a crystallographic point of view, this is not a primitive unit cell (UC), because a cubic face-centred UC never results in a cubic primitive UC. The primitive UC in this case is a rhombohedron with an opening angle of alpha = beta = gamma = 60°.
I don’t know how VASP etc. operates, maybe this ‘primtive UC’ as Avogadro handles it is exactly what VASP needs as input, but as I said, from a crystallographic point of view it is wrong to name this a primtive UC.
Hmm, this 1/8 ‘filled’ cell is neither a primtive nor a conventional cell, because the conventional cell is the case of NaCl the face-centered one. So for me, the question is indeed: what does the command do?
*Maybe the command should read as ‘Preparing the Unit Cell for solid-state quantum chemical calculations’?
Yes, that’s what I’m (and I guess a not too little part of the crystallographic community) asking for. You can of course ignore that but then Avo2 will not be the tool of choice for crystallographers. That’s completely ok.
The wording of the command as it is, is counterintuitive and what Avo2 does is, in my opinion, anti-crystallographically.
If the crystallographic section of Avo2 is only for preparing input files for other programs, I can live with that. But even at the risk of repeating myself, I would find it very unfortunate, if you can’t even make a proper image of a completely filled unit cell of NaCl that you can put for example on your website, make contributions to Wikipedia etc. Maybe we are from very different communities, but can you show me one example in which the fameous NaCl structure is shown like the way Avo2 does show it after the command in question? I have been involved in crystallography intensively for 15 years and saw this type of representation for the very first time in Avo2.
Yes, of course, but if you want your point of view to be consistently represented, then – as Thomas has shown – you also have to display half, quarter and eighth atoms instead of the redundant other halves, quarters, eighths…
Right. And I’m just trying to have a conversation about wording for different commands. Because Avogadro does serve multiple user communities. So we need to make it possible / easy to “fill” both with the translationally-equivalent copies that you want, as well as the “only symmetric positions, no translational duplicates” needed for DFT codes.
I asked the same thing last year, which was what is the most consistent wording?
I’m okay making “Fill Unit Cell” create the translational copies if that’s less confusing. I literally coded that for the little PNG graphics that go with the sample crystals. But then people complained that they had duplicate atoms in their DFT calculations.
I’m rambling. Clearly two commands are needed, but I cannot come up with good wording:
Fill Equivalent Positions (no translational equivalents)
Fill Unit Cell (including translational copies)
Other suggestions are certainly welcome. I’m happy if Avogadro has some use to crystallographers.
Perhaps following the convention in another program like XCrysDen would be good. The documentation there lists two (relevant) commands in the “Display” menu:
Unit Cell (with all atoms shown, including periodic replicas)
Translational Asymmetric Unit Cell (without periodic replicas, as something like Quantum ESPRESSO would use in an input)
I personally think these are good, if a bit wordy. I think I have access to Vesta through my work, so I can check at some point what their syntax is for that.
This is something that makes the whole problem very tricky. Once you start needing to export a crystal to a file type, Avogadro needs to be able to know exactly what the user wants to export. I personally am in favor of Avogadro only ever exporting a periodic version of a cell, and not a “full” cell. The periodic version is easily the more universal format, and is far more likely to be readable by another program. It also has the added benefit of not specifying redundant atoms, therefore reducing file sizes.
I think that I generally agree with @kohaerenz on what Avogadro should display, but I also think that it can introduce some unexpected behavior for users who aren’t intimately familiar with crystallography and condensed matter programs.
I can also add tooltips, which might be useful for some of these?
I’d be curious certainly. I’m happy to make “Fill Unit Cell” the complete version if someone can come up with better wording for the “Fill without translational equivalents”
Maybe “Fill Translational Unit”?
I started a pull request, which also adds a way to filter and search the space groups (which has bothered me for a while). I’ll think a bit more about the heuristics when importing a CIF.
As well as the two options for treatment of edge atoms discussed so far:
Show only the original atoms
Automatically duplicate the edge atoms so that they appear at the other edges, thereby changing the coordinates list/file
As I’ve mentioned in the past, I feel that there’s a third option too:
Show the implied atoms at the other edges without actually adding them
For what it’s worth, I think that would provide the nicest and most intuitive UI. It can then function as a viewing mode as opposed to a command that permanently changes the file contents.
What I mean is a mode where atoms on the edges are shown repeatedly but where the duplicate atoms are not actually added to the coordinates list, and all atoms that are actually the same atom behave as one e.g. changing the element of any of them changes the element of the actual true atom. As in, the atom exists once but appears in multiple places at the same time.
In a really ideal world the atoms would just wrap so that half the atom was on one side and half on the other, but it’s been discussed previously why that’s a challenge. I think it would work fine without that anyway though, just by displaying whole atoms.
Except the current rendering system is heavily optimized to show things that are actual atoms. That’s why, for example, it’s currently a pain to show extra unit cells without generating a unit cell.
It’s a nice feature request (i.e., show things that aren’t actual atoms) but it’s not something I can get to anytime soon.
Partly I guess I’m saying it in support of you/the current default being correct. In fact, I would argue against ever adding extra atoms automatically, even based on heuristics, if those atoms aren’t just fictitious reflections like the ones I described but are actually “proper”, independent atoms. I think it’d be more confusing if simply opening a file and then saving it would result in additional atoms being created without a explicit request by the user to do so. And like you said, it could cause a problem for some consumers of generated files.
So, given the technical challenges you mentioned that mean that any extra added atoms must for now be “proper” ones, I’d be more in favour of just making it as easy and as obvious as possible for the user to add (and remove?) the extra atoms to get the three different patterns/styles of cell that are wanted (primitive, conventional, and with duplicated edge atoms).
For a start that means coming up with really clear descriptions for the commands, because clearly they’re a bit lacking and prone to being misunderstood right now, but I don’t have good suggestions I’m afraid.
I think the most that I’d find sensible in terms of “automatic” filling of the cell would be a dialog that pops up when a crystal structure is opened asking which representation the user wants. If that would be too confusing for students/non-crystallographers, a brief recommendation could be made in the dialog for whatever is deemed most intuitive. If that would be annoying for people who work regularly with repeating structures then there could be an option to save the preference.
Basically I, like you, have some qualms about adding atoms that aren’t in the original file.
I think I’ll slowly say goodbye to this thread, I can’t contribute much more, but I still have one question: What do you mean by ‘adding atoms that aren’t in the original file’? In the CIF of NaCl, there are only two atoms specified, the asymmetric unit. All other positions in the cell are generated by the symmetry operations of the space group. Also the positions that the command ‘Fill Unit Cell…’ generates. Where is the difference then? Why do you allow symmetry operations that are not pure translations to add atoms but not translational equivalents? From a crystallographic point of view there is no difference between these two kinds of symmetry operations, and and it could be that our dialogue suffers a little from the fact that I argue crystallographically, while the programme performs specific tasks, e.g., creating “unit cells” that are neither primitive nor filled conventional, but are suitable for quantum chemistry programmes.
I think that’s the take-home message. You’re saying as a crystallographer that you want to see all the positions, including translationally equivalent ones.
Other users want “all the symmetric positions, but not the translationally equivalent ones”
I have a patch that adds both “fill” options as well as a heuristic when opening files:
Fill Unit Cell (all positions)
Fill Translational Unit (no translationally equivalent ones)
It also cleans up space group symbols, although getting the overline is driving me nuts. (Seems like Apple doesn’t include e.g. \overline{3} in their fonts.
Yes, it would be nice to have “render in extra positions” as a feature or showing clipped spheres, but those are a longer-term feature request. (Same with ellipsoids.)