Present day Avogadro 1.102.1 has the side panel display types with an entry Ball and Stick which, by tap on the later, opens its own sliders and radio buttons the program remembers for future use. By similar token, the entry Crystal Lattice could be amended with display options once set and kept until additional intervention by the user. Influenced by CCDC’s Mercury, entries here could be:
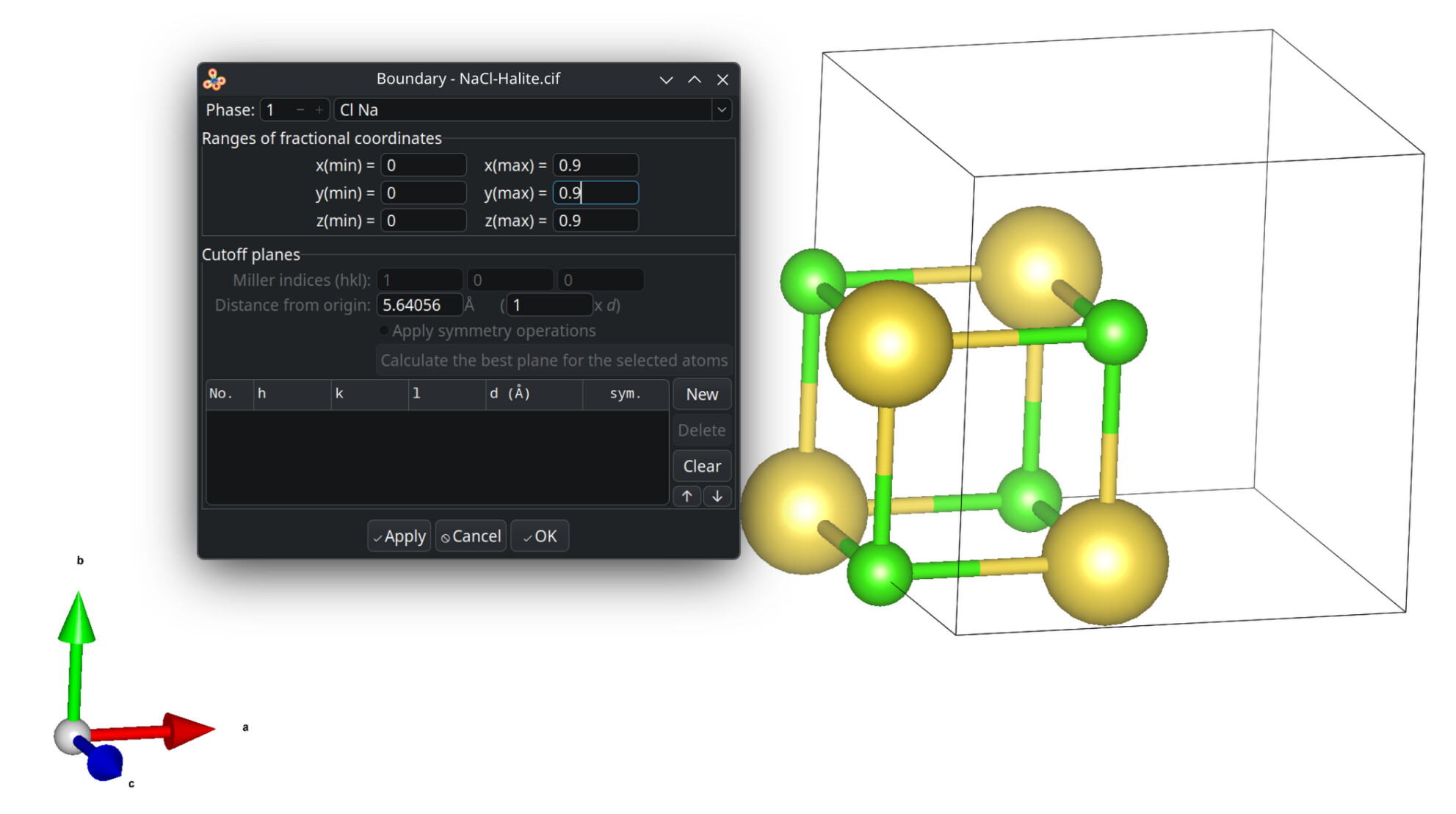
[ ] packing of a unit cell with
[ ] atoms which fit
[ ] atoms of molecules whose centroids fit
[ ] atoms of molecules where any atoms fit
[ ] atoms of molecules where all atoms fit
[ ] display of the asymmetric unit
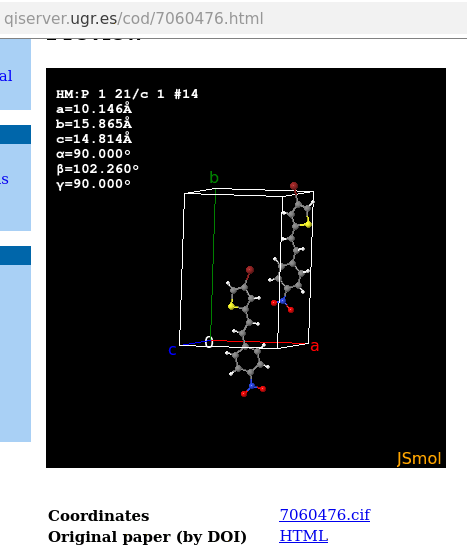
while Avogadro would default to the representation of the whole molecule. Here, “whole molecule” might require a visual completion of the representation for instance if asymmetric unit and symmetry operator (e.g., centre of inversion, axis of rotation) yields a molecule as one would write into a .sdf file. This is what e.g., codcif2sdf of cod-tools (DebiChem package, GitHub repository) provides, (including bond orders, which are not a requirement in .cif files). If a molecules appears to reach beyond the borders of the parallelepiped (example COD7060476)
codcif2sdf “reconstructs” as few sane molecules as deemed necessary for the .sdf (for this particular model in P2_1/c, one expects only one molecule in the .sdf). Note there is some overlap of cod-tool’s codcif2sdf and complementary cif_molecule, too.
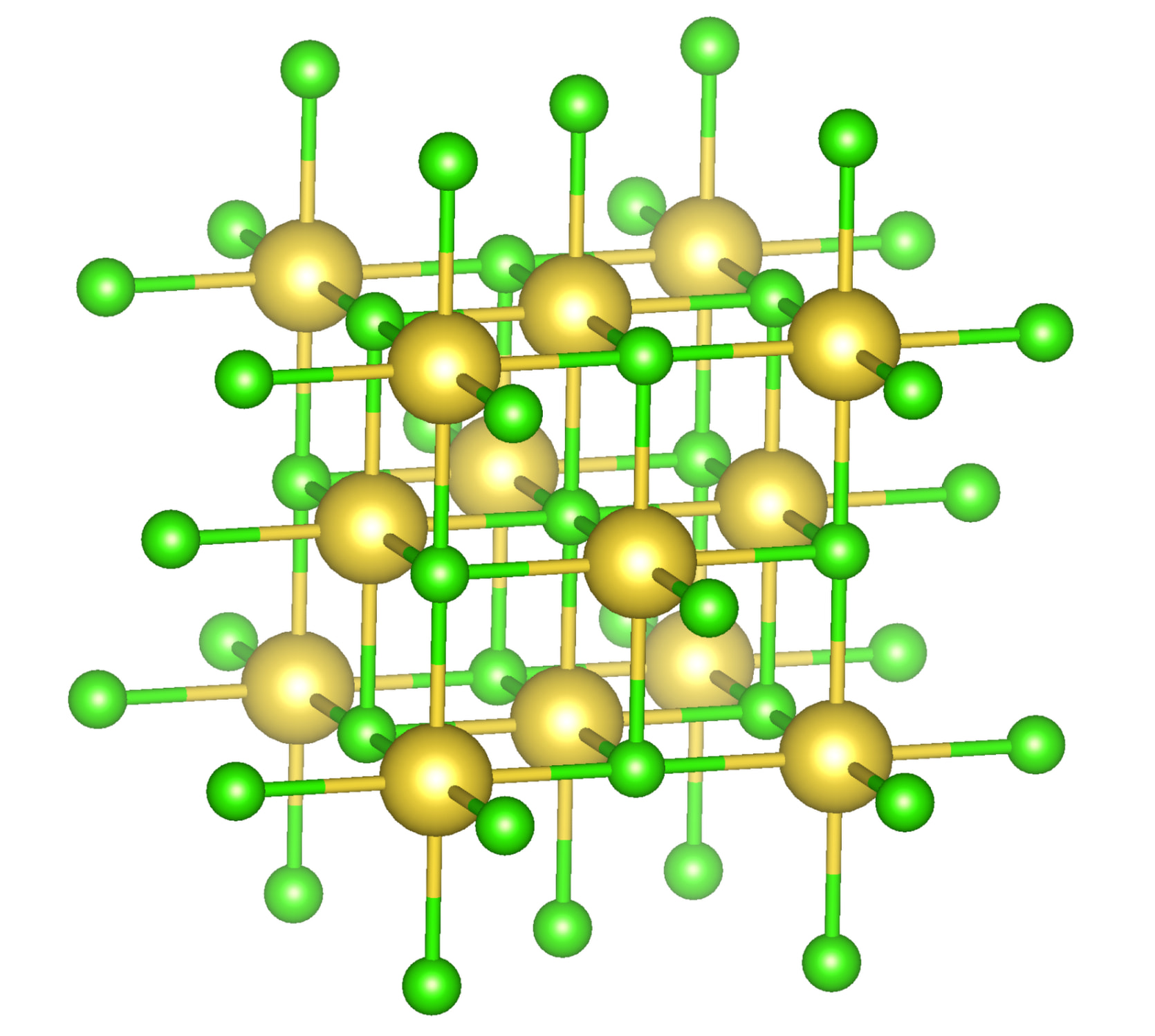
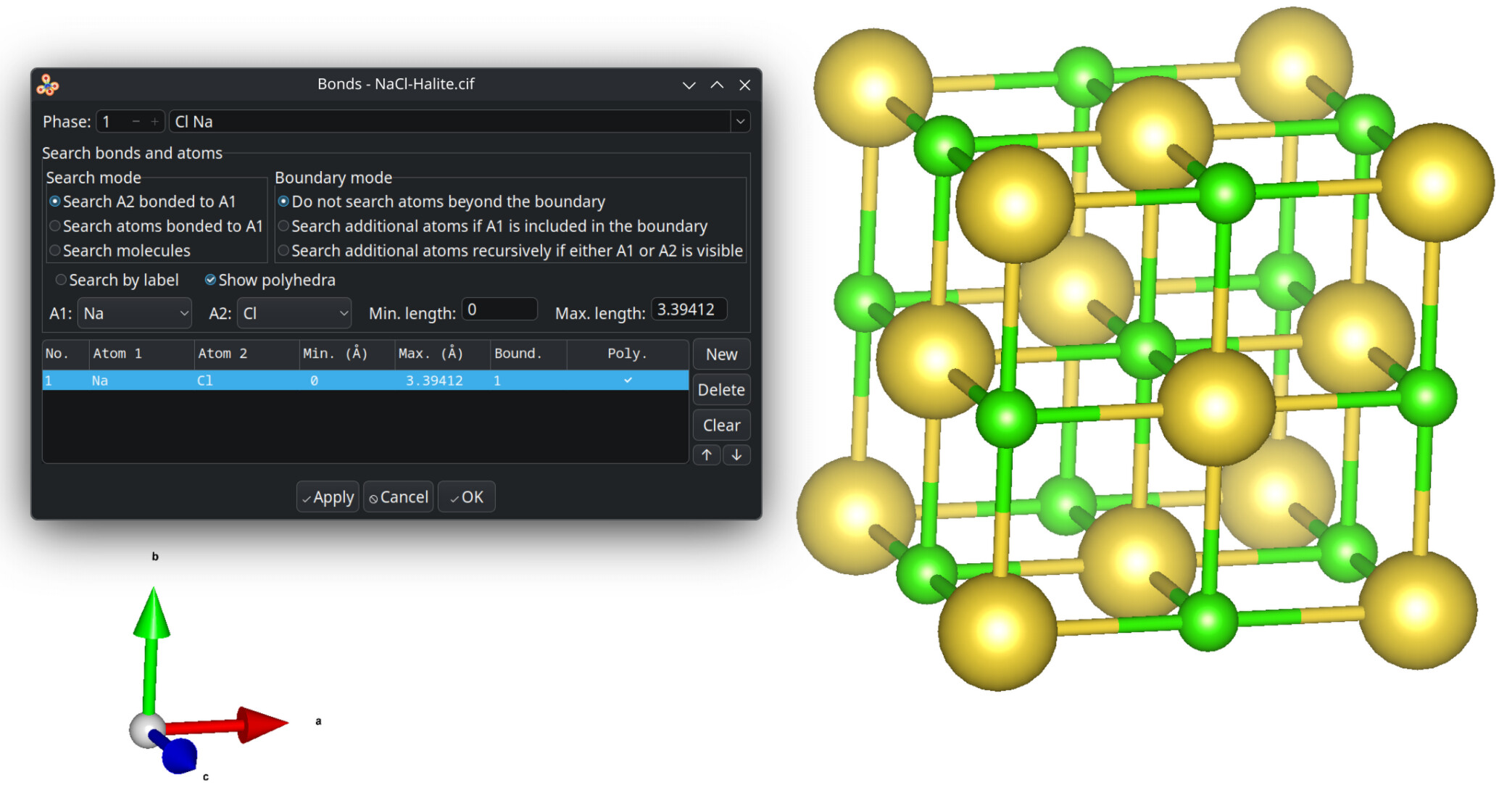
Within the suggested packing options, the four levels equally shall mutually be exclusive of each other. The preferred default is to include atoms of molecules whose centroids fit. Avogadro’s current “fill cell” is closer to cod-tools’ cif_fillcell (or Jmol’s load input.cif {1 1 1}) to consider atoms which fit at all the parallelepiped. Thus a subsequent search for bonds by Avogadro is affected, too.
I didn’t venture out cod-tools’s scripts and potential interdependence to account for. I don’t expect Avogadro 2.0 to fill each of these desiderata, too.
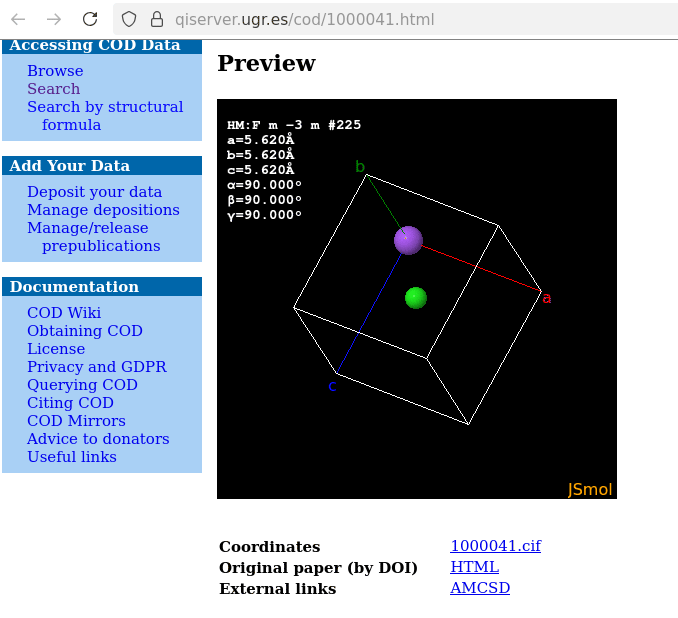
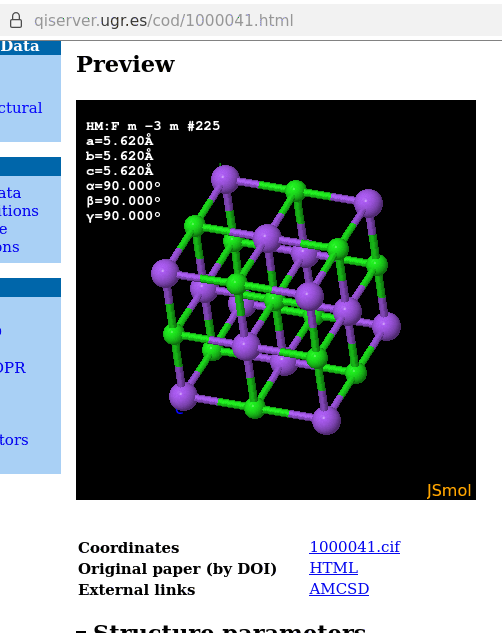
A note on / about sodium chloride: the default display with JSmol of the corresponding model equally displays the unit cell virtually partially filled:
which is the result of the database’s interface running a load cif/1/00/00/1000041.cif {1,1,1}; hide symmetry instead of a load cif/1/00/00/1000041.cif {1,1,1}; on the website’s console:
A note on Jmol: the different display of e.g. load input.cif; vs for instance a load input.cif {1 1 1} equally affects the generation of an input file for Gaussian (tools → Gaussian).