I recently have been opening a large number of .cif files for my research, and I noticed that often there is a section of the listed coordinates that is part of a “disorder” group, which arises from long range disorder in the crystal structure. This syntax does not seem to be known to the file reader that Av2 uses, I believe it is from OpenBabel. The fix would be writing a few lines into cifformat.cpp that checks for the tag _atom_site_disorder_assembly or _atom_site_disorder_group in the loop section of the .cif file, shown here:
After detecting that there is disorder present, then it is a simple matter of just picking the atoms from a single disorder group, which you can see are distinguished with the following notation:
C2 C 0.0781(6) 0.5708(6) 0.5617(4) 0.170(5) Uani 0.897(6) 1 d . . P B 1
H2A H 0.070259 0.574604 0.500206 0.255 Uiso 0.897(6) 1 calc R U P B 1
H2B H 0.042938 0.605123 0.577230 0.255 Uiso 0.897(6) 1 calc R U P B 1
H2C H 0.043126 0.533561 0.568850 0.255 Uiso 0.897(6) 1 calc R U P B 1
C3 C 0.2459(4) 0.5611(3) 0.7580(3) 0.0623(15) Uani 0.897(6) 1 d . . P B 1
H3A H 0.214148 0.522990 0.765911 0.094 Uiso 0.897(6) 1 calc R U P B 1
H3B H 0.208713 0.594415 0.773728 0.094 Uiso 0.897(6) 1 calc R U P B 1
H3C H 0.324135 0.561951 0.796252 0.094 Uiso 0.897(6) 1 calc R U P B 1
C4 C 0.2853(10) 0.4994(2) 0.6091(5) 0.170(5) Uani 0.897(6) 1 d . . P B 1
H4A H 0.284467 0.502801 0.549455 0.255 Uiso 0.897(6) 1 calc R U P B 1
H4B H 0.240816 0.464912 0.611650 0.255 Uiso 0.897(6) 1 calc R U P B 1
H4C H 0.360823 0.493805 0.651745 0.255 Uiso 0.897(6) 1 calc R U P B 1
Si1 Si 0.22789(17) 0.56917(13) 0.63652(8) 0.0528(6) Uani 0.897(6) 1 d . . P B 1
C3A C 0.212(4) 0.607(3) 0.759(2) 0.0623(15) Uani 0.103(6) 1 d . . P B 2
H3A1 H 0.290939 0.609112 0.796619 0.094 Uiso 0.103(6) 1 calc R U P B 2
H3A2 H 0.179965 0.572719 0.777442 0.094 Uiso 0.103(6) 1 calc R U P B 2
H3A3 H 0.176749 0.644143 0.766347 0.094 Uiso 0.103(6) 1 calc R U P B 2
C2A C 0.065(5) 0.624(5) 0.592(4) 0.170(5) Uani 0.103(6) 1 d . . P B 2
H2A1 H 0.053638 0.656635 0.627572 0.255 Uiso 0.103(6) 1 calc R U P B 2
H2A2 H 0.013368 0.591386 0.587216 0.255 Uiso 0.103(6) 1 calc R U P B 2
H2A3 H 0.053501 0.638871 0.533298 0.255 Uiso 0.103(6) 1 calc R U P B 2
Si1A Si 0.1929(11) 0.5982(7) 0.6416(6) 0.036(3) Uani 0.103(6) 1 d . . P B 2
The problem next is to implement something into the Av2 UI that allows someone to pick which disorder group to display in the crystal. I think the best thing for that would be to include another section in the Crystal menu for picking the disorder group, which might be something I work on if I can find the time.
I’m not that familiar with disorder groups. If I understand, the CIF is essentially noting that there are different coordinate sets for the atoms because of disorder.
If so, that’s not too hard to add to Open Babel - basically, you’d note the disorder group and create multiple coordinate sets. I guess one question - is it always going to be all atoms? Or do you need to track that some atoms are in a disorder group, but others will have identical coordinates in each set.
Anyway, once that’s added to the Open Babel code, it’ll come through cleanly into Avogadro.
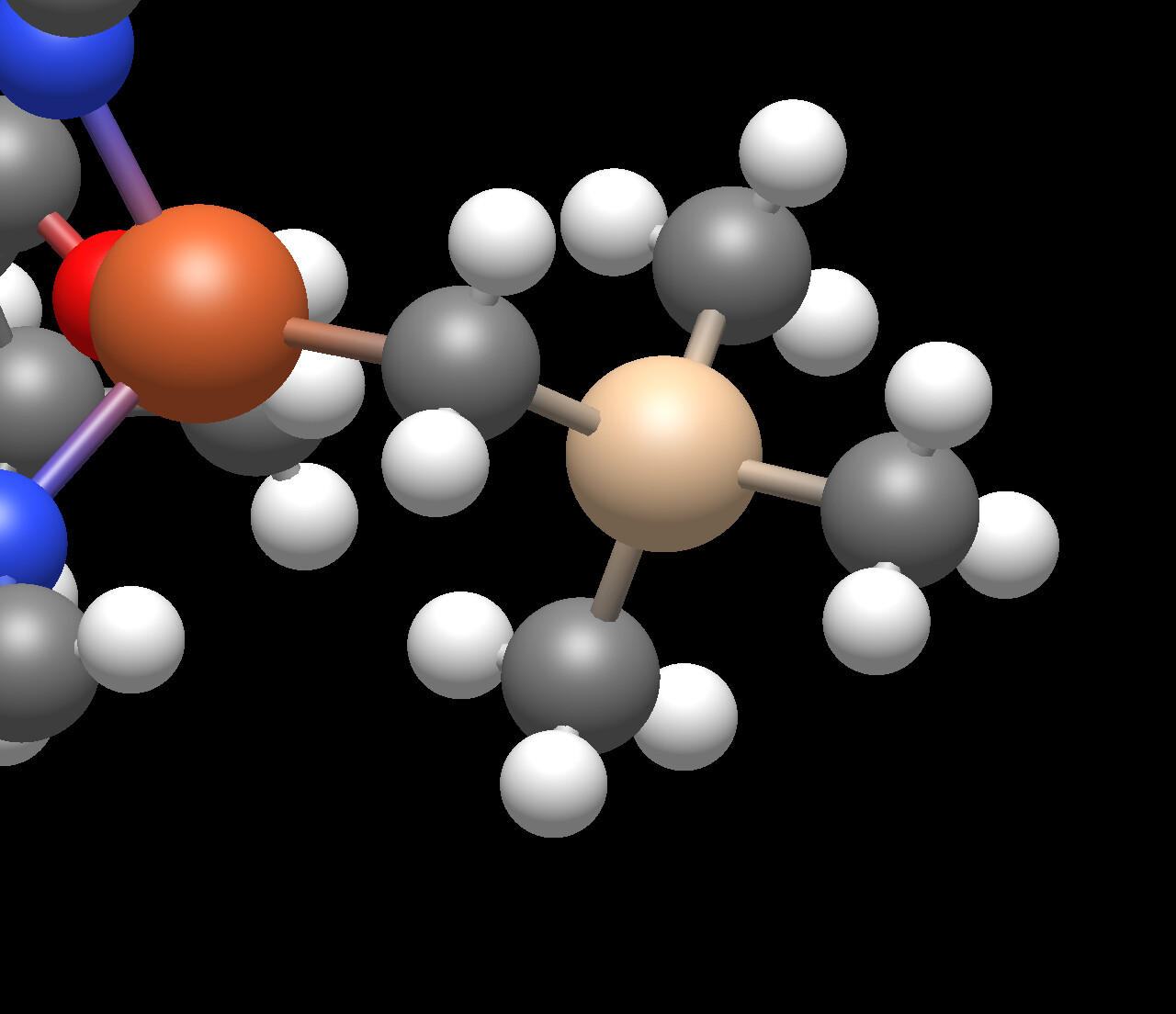
From what I can tell it isn’t always all the atoms, though I imagine it could be all of them. The example I sent was from a much larger crystal structure, and only part of the tetramethylsilyl group had the disorder group. I will take a look at the code and try to figure out how to add it, but to be honest these days I am still just trying to learn C++.
I recall that in the past you have held workshops for Av2 development, perhaps it would be a good time to have another, given that there seems to be some more energy in the development space (mainly me and @matterhorn103 but still that’s more than the Geoff solo adventure that has been happening for a while).
@brockdyer03 It more likely was a -SiMe3 group, rather than a tetrametylsilyl group (an other typical candidate are the terminal -CF3 and (in principle) -CH3 group*, like a piano stool). Disorder then can extend to whole molecules “at the switch” between space group P2(1)/c to P2(1) to yield orientational disorder in polar materials.
But the example shared displays the underlying principle in a .cif file. Presumed there are only two (sets of) atomic positions, there is one probability p_1 for one set, and the other is p_2 = 1 - p_1. Disorder equally can be occupational if for instance in some derivatives of Prussian blue where the same site (i.e. position in the crystal) can be used by one ion (probability p_1), or an other (p_2, same mathematics as above).
There are programs out there to display, and allow to split disorder for the subsequent separate computation of intermolecular interactions. CrystalExplorer is an example (see here). On occasion crystallographic disorder shows up in the user list of Jmol (example, example) since the program can be used to display structures, and prepare a subsequent computation with Gaussian. If Avogadro wants to enter this field, is there an exchange with the authors of either one of the two other programs?
* But in comparison to -CF3, the -CH3 often is not modeled in positional disorder if the diffraction experiment is with X-rays “only”. In neutron diffraction, the signal caused by hydrogens typically is much better/good enough to model disorder of a terminal -CH3 (if there is enough evidence).
I am aware of the programs you mentioned that would be able to do this, and while I am sure there are other authors who would share their work, it is probably for the best that we maintain our own code.
I think the main thing in this case is if you (or @Thomas) can share a few example CIF files. In principle, it shouldn’t take too long to implement in the CIF reader. Something similar is done in PDB files, for example.
I can either set aside some time, or show some example code for an Open Babel patch – whichever you prefer.
While I intend on scheduling another “Avogadro User’s Meeting,” the easiest is to offer mentorship pointers and “good first issue” examples. Everyone has slightly different interests, backgrounds and expertise, so it’s often hard to have a one-size-fits-all session.
@brockdyer03 Can you share the complete .cif file (which would allow to walk around the whole structure)? Because in the projection shared by you, I recognize three methyl groups (\ce{-CH3}) and one bridge which looks like a \ce{-CH2{}-}methylene group.
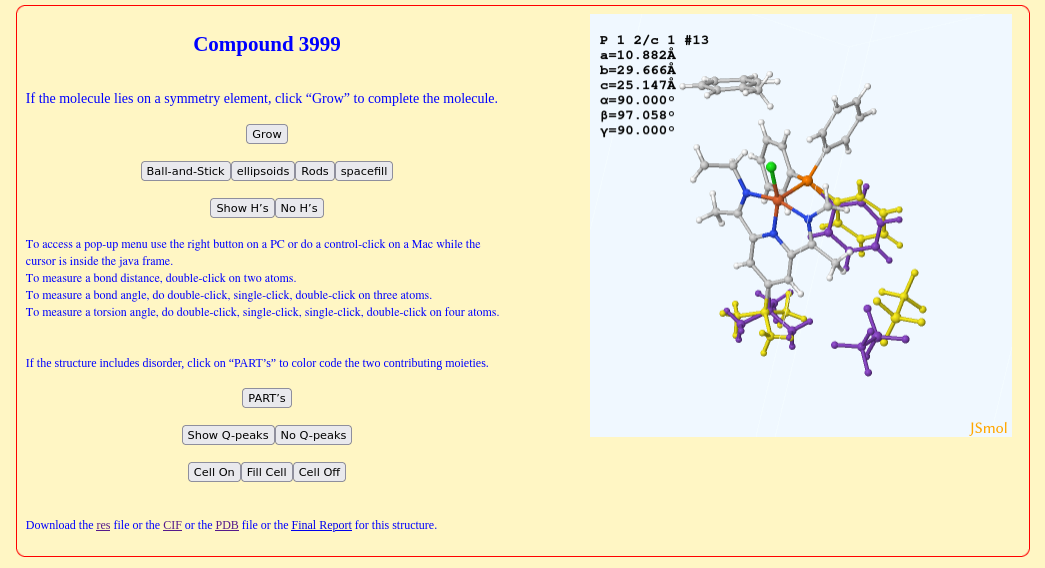
In a recent discussion about disorder and its display in Jmol, Patt Carroll (UPenn) recently hinted “compound 3999” (link) positional disorder of a phenyl ring, tBu group and ethane (highlighted in the non-CPK color scheme):
Had to rename it since the .cif file extension isn’t allowed to be uploaded here for some reason. Regardless of what the name of the group is, there are 4 carbons attached to a silicon, and tetramethylsilyl gets the point across. cs2c05838_si_002.txt (2.5 MB)
I recently switched from Avogadro 1 to Avogadro 2 (v1.100.0) and I noticed that cif file formats are not available for opening a file or saving the file. I find it weird, so I was wondering if I am missing some dependency or installed a beta version that is why. I installed on windows using x64 .exe installer.
There is a known bug with 1.100 - it did not include Open Babel for opening file formats like CIF. This seems to be fixed in the nightly build - but we are not sure yet why the release script did not work correctly.