@MariaLidia Based on the screen photo you shared, you appear to use Avogadro of the old (and no longer actively maintained) branch of Avogadro (because of the icon of two arrows next to the ruler icon). An example would be version 1.2.0:
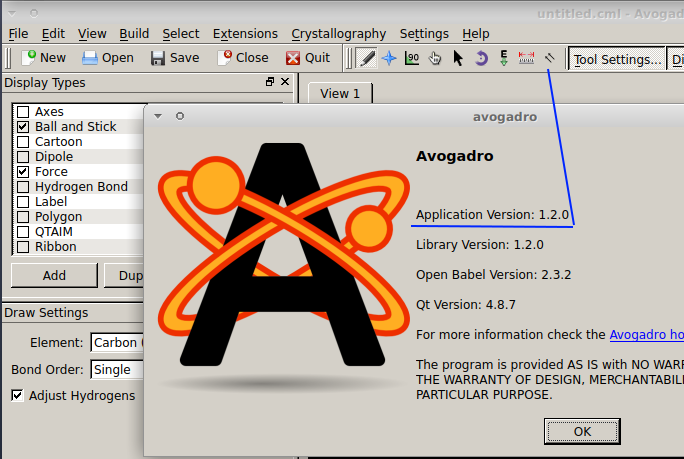
If your workflow permits, I recommend to ascertain a current version of OpenBabel (releases page) and Avogadro of the branch leading to Avogadro2
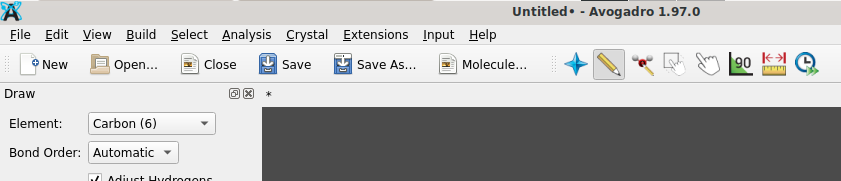
as provided on this landing page. If you like to continue to use Avogadro’s input generators for Gaussian, MOPAC, etc. you additionally need an installation of Python and to indicate the location of the Python interpreter (Extensions → Set Python Path), followed by their installation (Extensions → Download Plugins).
The geometry optimization is launched either by simultaneous touch of Ctrl + Alt + o, or with mouse click via Extensions → OpenBabel → Optimize Geometry.