Avogadro version: 1 2 0
Operating system and version: Windows 10
Hi, I’m a begginer in Avogadro so there’s probably a simple solution to my problem. When I build molecules and try to optimise them energetically, some random atoms aren’t affected by the force field at all. Let me illustrate this with an example.
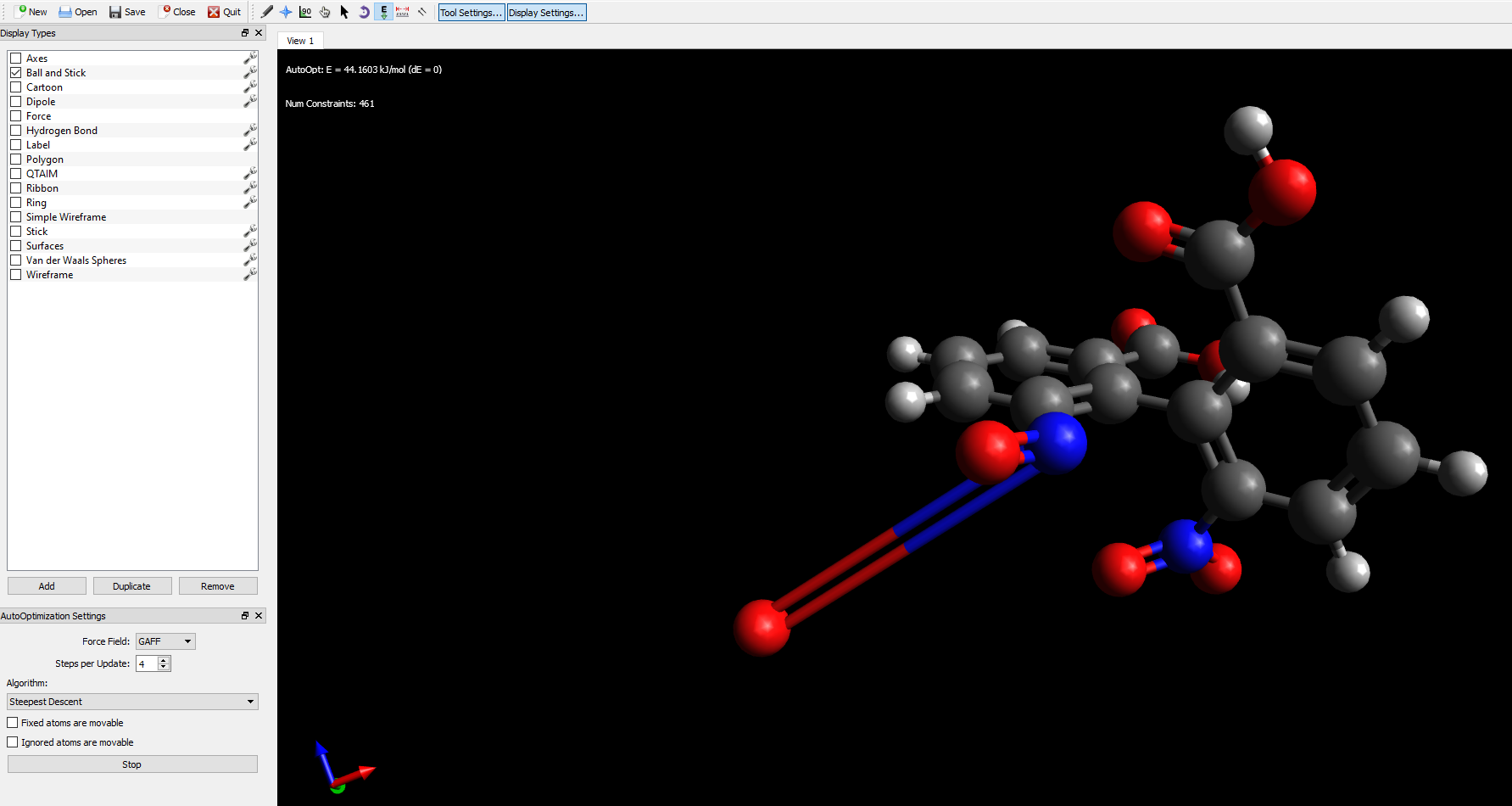
The case here is the oxygen atom of the nitro group. The bond is visibly elongated. This oxygen atom seems to be fixed in this one place. When the Auto Optimalization is running, it doesn’t respond to clicking and dragging, unlike other “normal” atoms- despite all my efforts is still unmovable. I can use the Manipulation Tool to a bit adjust its position, as I know what it probably looks like, but it isn’t suppoused to work like that.
It doesn’t matter what molecules I build, what atoms I use. This disease affects them randomly and I have no idea what the cure might be. Changing the force field doesn’t work either.
I’m sorry if I’m asking about something that was previously mentioned and I haven’t noticed it in the discussion topics.
In your screenshot, it says “Num Constraints = 461” which suggests that for whatever reason, there are optimization constraints on that atom.
My suggestion would be to save the file (e.g., to CML or SDF), close and re-open to optimize. The constraints will not be saved with the file, so when you re-open it should not happen.
I’m afraid it didn’t work. When re-opened, this N=O bond of the molecule turned out to be unchanged. And when I wanted to optimalize it, I got a comment: “AutoOpt: Could not set up force field…”.
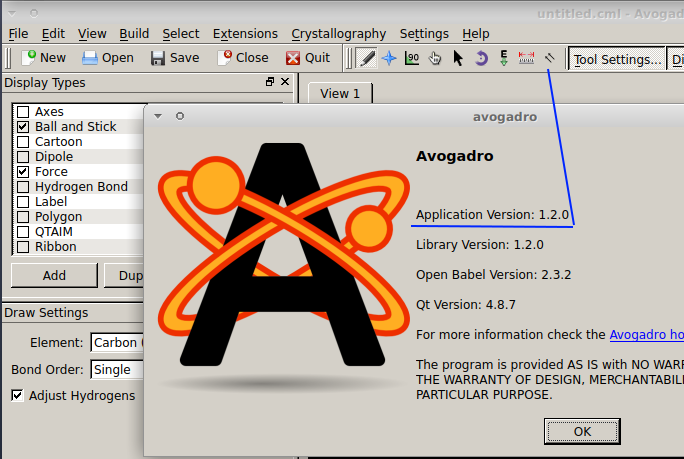
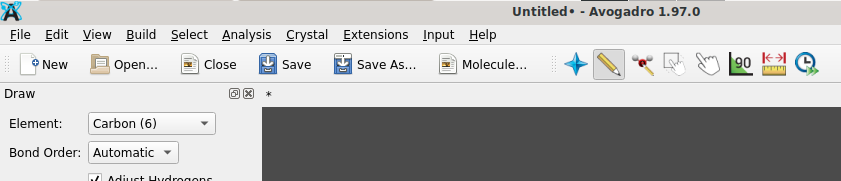
@MariaLidia Based on the screen photo you shared, you appear to use Avogadro of the old (and no longer actively maintained) branch of Avogadro (because of the icon of two arrows next to the ruler icon). An example would be version 1.2.0:
as provided on this landing page. If you like to continue to use Avogadro’s input generators for Gaussian, MOPAC, etc. you additionally need an installation of Python and to indicate the location of the Python interpreter (Extensions → Set Python Path), followed by their installation (Extensions → Download Plugins).
The geometry optimization is launched either by simultaneous touch of Ctrl + Alt + o, or with mouse click via Extensions → OpenBabel → Optimize Geometry.