I am using Avogadro 1.99.0 to create input for Orca. The molecule is purine(7H).
I cannot see a way to determine which atom is #1, which is #2, etc. This information appears not to be displayed or displayable, which makes analyzing the output difficult. For example, NBO analysis by ORCA running NBO7 reads:
Natural Population
Natural ---------------------------------------------
Atom No Charge Core Valence Rydberg Total
--------------------------------------------------------------------
C 1 0.00966 1.99998 3.96870 0.02166 5.99034
C 2 0.30955 1.99998 3.66054 0.02993 5.69045
N 3 -0.52457 1.99999 5.50553 0.01905 7.52457
N 4 -0.44693 1.99999 5.41201 0.03493 7.44693
N 5 -0.41695 1.99999 5.38374 0.03322 7.41695
C 6 0.23518 1.99999 3.73939 0.02545 5.76482
C 7 0.05162 1.99999 3.92429 0.02411 5.94838
N 8 -0.44795 1.99999 5.41567 0.03229 7.44795
C 9 0.22993 1.99999 3.73943 0.03066 5.77007
H 10 0.20368 0.00000 0.79494 0.00138 0.79632
H 11 0.41888 0.00000 0.57939 0.00172 0.58112
H 12 0.18178 0.00000 0.81663 0.00160 0.81822
H 13 0.19613 0.00000 0.80190 0.00198 0.80387
====================================================================
* Total * 0.00000 17.99988 43.74216 0.25796 62.00000
Obviously, Avogadro2 is not using the standard numbering for the purine ring system. So how do I determine which atom is which, in a non-error-prone way?
Note: I tried searching the Avogadro2 manual webpages for “atom numbering” and “atom labels” but I didn’t find the results helpful.
But you can also open up Analyze → Atom Properties…
When you click on an atom in the spreadsheet, you’ll see a particular atom selected.
If you wouldn’t mind posting the Orca output, it shouldn’t be too hard to parse the NBO output (e.g., the natural charges)
I don’t know what you mean by that. Avogadro is going to use the numbering of the atoms order in your input file. That would depend on how you created the purine in the first place.
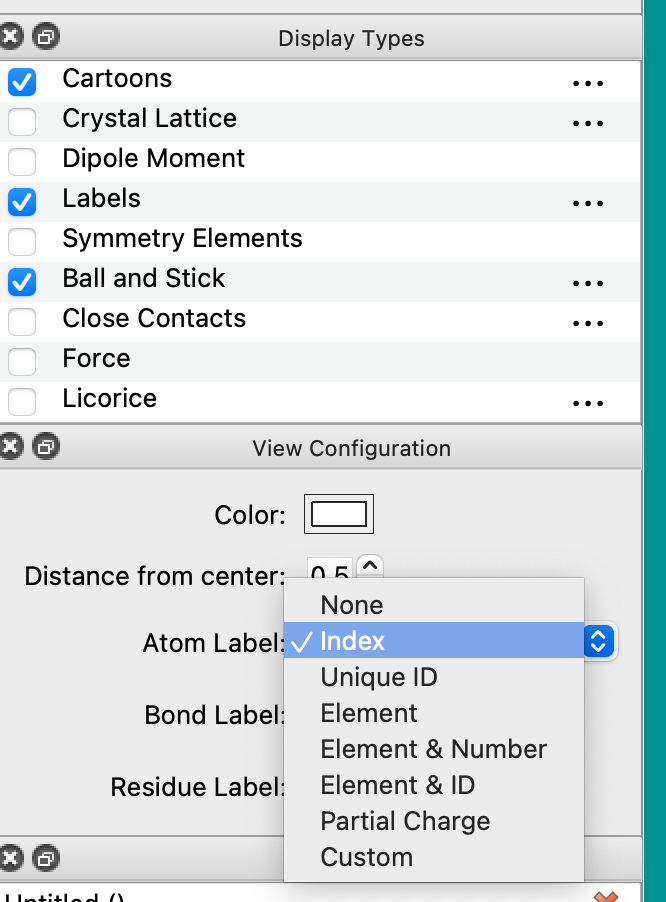
But yes, the “Labels” rendering gives atom numbering among other options.
Thanks. With your help, I did find the “Labels”, although it took a little trial and error to get it to display label numbers instead of symbols.
I don’t know what you mean by that. Avogadro is going to use the numbering of the atoms order in your input file. That would depend on how you created the purine in the first place.
Purine has an internationally approved numbering. That is what I meant. Not that I really expected Avogadro to use it, but I thought I might get lucky. My complaint is not that this numbering wasn’t used, but that I could not determine what the numbering was.
Of course, ORCA numbers the atoms using the order in the list of atoms, which is determined by Avogadro’s output.
As for how the purine molecule was generated, because I did not find purine itself as an insertable molecule, I inserted adenosine phosphate and deleted a bunch of atoms.
And of course, don’t forget to watch out for the fact that the NBO analysis uses indices starting at 1, whereas ORCA itself will start from 0!
In Avogadro 1.99, this corresponds to Atom Label options of Index (starting at 1) and Unique ID or Element & ID (both starting at 0). In the dialogs like Atom Properties it always starts at 1.
The sequence of atoms in an .xyz file needn’t to follow the numbering scheme of parent structures outlined in IUPAC’s Blue Book (links compiled by chemistry.stackexchange). The two are independent of each other. However it is true that files with more advanced syntax than .xyz, if they have a block or connectivity table (e.g., mol/sdf files) need to keep this part is “in sync” with the coordinates table.
As an example of illustration, use CC1=CC=CC(C)=N1 as input SMILES string currently displayed in the property box of 2,6-lutidine in the English edition of Wikipedia for a conversion by openbabel 3.1.1:
$ obabel -:"CC1=CC=CC(C)=N1" -h --gen3d -O file1.xyz
1 molecule converted
$ cat file1.xyz
17
C -2.42127 1.11845 0.01253
C -1.15964 0.30889 0.01358
C -1.20009 -1.08050 0.00901
C -0.00098 -1.78390 0.00200
C 1.20035 -1.08442 0.00112
C 1.16445 0.30523 0.01071
C 2.42827 1.11084 0.02328
N 0.00367 1.00421 0.01455
H -3.31229 0.48341 0.02637
H -2.46075 1.74218 -0.88589
H -2.44880 1.76488 0.89496
H -2.14556 -1.61259 0.01141
H -0.00257 -2.86971 -0.00252
H 2.14439 -1.61920 -0.00448
H 3.31746 0.47239 0.02141
H 2.45967 1.73687 0.92026
H 2.46806 1.75474 -0.86106
where nitrogen is the last non-H atom prior to the hydrogens, atom index 8. Or expressed by an other, equally valid SMILES string which happens to be the one OpenBabel provides as the canonical SMILES string of the above, Cc1cccc(n1)C, i.e.
$ obabel -:"CC1=CC=CC(C)=N1" -ocan | obabel -ismi -h --gen3d -O file2.xyz
1 molecule converted
1 molecule converted
$ cat file2.xyz
17
C -2.42116 1.11930 0.01208
C -1.16049 0.30927 0.01306
C -1.20142 -1.08037 0.01099
C -0.00156 -1.78374 0.00183
C 1.20024 -1.08448 -0.00112
C 1.16429 0.30517 0.00883
N 0.00300 1.00324 0.01298
C 2.42751 1.11054 0.02371
H -3.31278 0.48421 0.02468
H -2.45918 1.74374 -0.88558
H -2.44879 1.76467 0.89546
H -2.14652 -1.61257 0.01600
H -0.00357 -2.86975 -0.00171
H 2.14368 -1.61988 -0.00827
H 3.31697 0.47268 0.02333
H 2.45680 1.73563 0.92162
H 2.46868 1.75551 -0.85944
where the nitrogen atom is the penultimate (second to last) non-H atom, atom index 7. If you fetch the .sdf from NIH-CACTUS, nitrogen happens to be very first atom in sequence of the .mol file (cf. the attached .tar.gz archive). This shouldn’t affect the structrue2name functions as provided by BioVia Draw, Chemsketch, Chemdoodle, ChemDraw, etc.