A colleague asked me for some example files for teaching VSEPR (eg 5-coordinate cases, SF4, 6-coordinate etc)
Any interest in posting these as part of the teaching examples?
A colleague asked me for some example files for teaching VSEPR (eg 5-coordinate cases, SF4, 6-coordinate etc)
Any interest in posting these as part of the teaching examples?
I had a lab in my first chemistry course that used Spartan to calculate geometries for a few small molecules, with the specific aim to examine VSEPR theory in it.
Here’s the list of 10 molecules that we looked at. I’ll leave figuring out the correct geometries as an exercise for the reader ![]()
BrF5.cjson (2.7 KB)
SOF4.cjson (2.7 KB)
ICl4-.cjson (2.5 KB)
SF4.cjson (2.5 KB)
BrF3.cjson (2.6 KB)
CH2O.cjson (2.6 KB)
OF2.cjson (2.4 KB)
PCl3.cjson (2.6 KB)
SiCl4.cjson (2.7 KB)
H2O.cjson (2.4 KB)
Interesting. A few of these were also on the list that Sean asked. The various triatomics (e.g., \ce{H2O} vs. \ce{H2S} and \ce{OF2}) are also good examples.
Seems like it’s probably worth getting a few into the library and writing up an example lab exercise. (Another colleague is working with a student on a lab exercise that will also include square planar vs. tetrahedral methane using GFN2 .. and we’ve talked about comparing 5-coordinate species since that always seems to be confusing.)
But it’s been since grad school that I’ve taught first-year chemistry, so I welcome feedback.
I still have a copy of the lab exercise that my professor wrote. If you’d like I can request permission from him to share a copy with you that might help guide the creation of a new lab exercise.
On a related note, I’d be a bit cautious about the level of theory used for the lab. For example, all of the structures I provided were optimized with GFN2-xTB, but upon closer inspection (and me finding the notes I took on that lab), not all of the structures were completely accurate.
For example, the one anion, \ce{ICl4-}, had the incorrect structure due to the how I drew and optimized the molecule. The structure there should’ve been square planar, but instead was a strange combination of octahedral electron geometry and seesaw molecular geometry, which really shouldn’t happen. In addition, I noticed the structure of \ce{SOF4} seems to be incorrect according to my notes from that class. Now granted, I was a freshman and took notes about as well as a mushy pear, so I may have made some mistakes then.
In that spirit, I’m rerunning all of the geometry optimizations with some nicer methods, in particular wB97M-V/def2-TZVPP. Once I get all of the structures re-optimized at that level of theory I can provide a more definitive set of geometries for all of the molecules, and I can confer with my prof to see if he agrees with the geometries I came up with.
The trick of course is that some of the other geometries are local minima. For example, you can often get a 5-coordinate species to take a square pyramidal structure as an intermediate, and while \ce{SF4} is see-saw, it can also be trapped an a higher-energy tetrahedral shape.
(In general, the 5-coordinate square-pyramidal geometry is a transition state in the Berry pseudorotation but in some compounds you can optimize it as a local minima as well.)
In general, I found even with GFN2, it got the relative energies correct, so comparing tetrahedral \ce{SF4} and see-saw energies is a useful exercise (i.e., see-saw wins).
If you’re willing to run a few ωB97M-V/def2-TZVPP optimizations, that would be great.
I optimized each molecule with ωB97M-V/def2-TZVPP and VeryTightSCF/VeryTightOpt in ORCA.
Tragically, \ce{ICl4-} shows up as not being square planar with those settings, it’s deviated by a reasonable amount.
I’m not 100% sure why, but at any rate I’ve decided that I don’t really want to spend the time and energy towards finding out why, so instead of that I’ve just included the optimizations with normal convergence criteria, which gave the expected geometries (for the most part).
VSEPR_Out.tar.gz (7.1 MB)
I haven’t run the calculations yet, but I know some standard examples I have seen before (text books ect.) I just put down the once that weren’t mentioned yet.
(These might already be included by your colleague)
NH3
CH4
CH3F
BH3
BF3
Then some more exotic ones
IF7
IF8^{-1}
XeF8^{-2}
Would it also be pedagogically useful to include, at the end, a couple of examples where VSEPR breaks down and no longer predicts the correct structure?
Not with the idea that the student should be asked to rationalize them, or even necessarily that the guide would rationalize them, but just to demonstrate that it has its limits as a theory, which I think is always good at an introductory chemistry level.
Looking at Wikipedia, famous cases seem to be XeF6 (octahedral not pyramidal) and Li2O (linear not bent).
I’m going to leave that to colleagues who have thought about and taught intro chemistry more consistently.
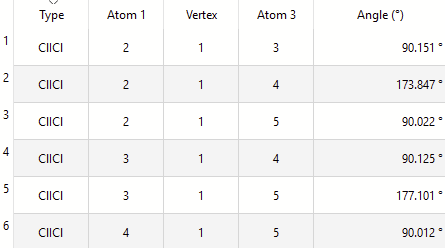
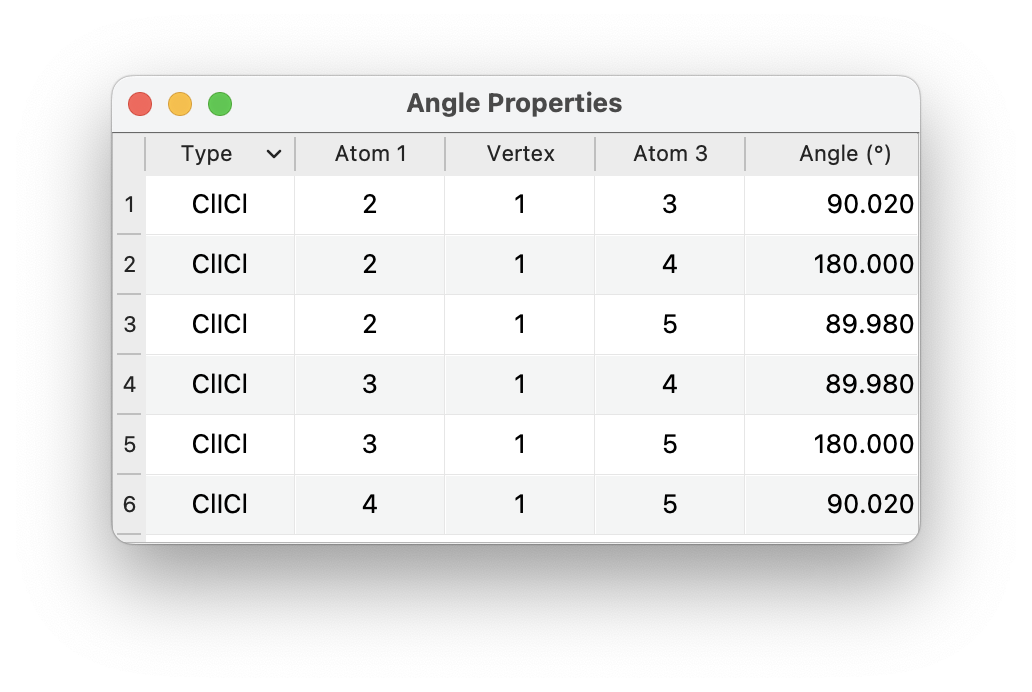
I looked at the \ce{ICl4-} case. With default “normal” tolerances, Avogadro (libmsym) finds a D_{4h} symmetry as expected. (Not sure why some of the angles are showing 173° .. maybe your molecule isn’t perfectly flat?)
I suspect the initial input wasn’t quite symmetric? How did you create the input for it? (Just want to make sure there isn’t a problem with the square planar template, for example.)
Here’s my output:
ICl4-.out (167.8 KB)