Maybe this is very simple but I can’t figure it out for the life of me. It’s a very similar question to this one.
I have an orca output file which I converted to the molden format with orca_2mkl basename -molden. I’m able to visualize the orbitals but when I select the density in the Create Surface menu, it just doesn’t do anything. I’m on Avogadro 2, Avogadro 1 even crashes when I try that or loads infinitely. Using fchk files works to create a density, so it is probably not an issue with the density plotter. I tried other molden files that weren’t converted but they don’t work either.
The AppImage from here for Linux (i.e., version 1.95 of Avogadro), equally fails to read the file.
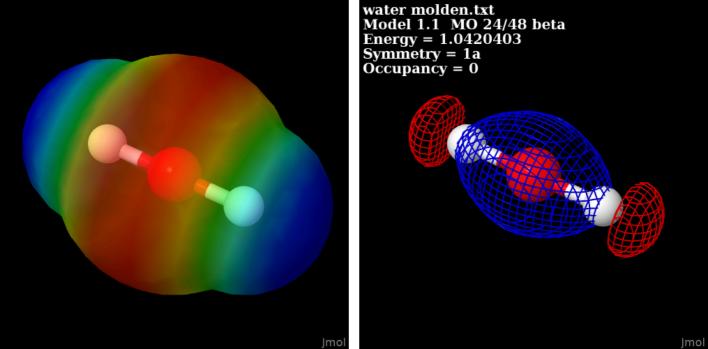
My next guess was to attempt reading the file with Jmol. Though neither orca, nor molden is explicitly mentioned in their compilation of supported file formats as input, the geometry is read promptly. (What looks similar to point group symmetry D_\infty{}h, like in acetylene, is unusual; possibly you skipped a preliminary geometry optimization with a force field like UFF, or MMFF96 before engaging the computation whose results you submit.) It seems noteworthy, if you read the file molden file from Jmol’s console (large portions of the program’s actions are easier accessed by scripting [File → Console], than the GUI) with
load "water molden.txt";
Jmol reports the warning: Unsupported orbital type ignored: [9G].
Nevertheless, from Jmol’s canvas, right-hand mouse click Surfaces → MEP, or Surfaces → Molecular Orbitals → 24 to visualize orbital #24 works, though again, note the symmetry of the molecule merits revision, or (if it is a snap shot of a vibration) justification:
Thanks for your answer and for checking it with Jmol. I’m aware that the geometry is funny, but I’m sure this doesn’t matter for the program whether it’s possible to represent densities.
The orbitals also work in Avogadro2, so I it’s not surprising that it also works in Jmol, especially since they list molden here.
In any case, we have to figure out what’s wrong with the file or Avogadro, because I need the density visualized in Avogadro
I wasn’t aware about the existence of the second compilation. What I like about the one I indicated is that it includes links to a library of typical files, deposit on sourceforge. And actually, there is one about still (like in non-vibrating) water by Molden: water_novib.molden
The comparison of this one with the file filed by you yields a few differences. Do you have access to a diff-viewer like kdiff3, or meld to spot similarities and differences of two text files? Doing so, for example, reveals blank lines initiated by [5D], [7F], and [9G] in your file do not exist in the example for Jmol. On the other hand, Jmol’s file contains e.g., a block initiatialized by [GEOCONF] absent in the translated Molden file you submitted here.
However, with the AppImage 1.95 of Avogadro for Linux and Jmol’s reference file about Molden in in hand, I do not reach further than reading the geometry successfully. Analysis → Create Surfaces, works as well as the choice for a MO like the LUMO, or electrostatic density. Once I click on “calculate”, Avogadro briefly displays a progress bar, then however disappears for good. Based on this observation, I speculate Avogadro has some problems reading the wanted information.
Looking through the code, I’m not sure that Avo2 will generate the density matrix from the orbitals if it’s not reported. (I’m adding that to the TODO)
For other reasons, I wanted to do this with Python and it’s fairly easy to do with Orbkit:
From a command-line: orbkit -i file.molden.gz --otype=cb --adjust_grid=5 0.2 -p 4 # generate a cube file for Avogadro, etc.
Oh no! I was already working a bunch inside here. I think I assumed that since there is code to build the density, it would also be possible to display it?
I can’t estimate how hard or easy it is to have that functionality to show the electron density surface or whether it makes sense to help?
EDIT: From what I see, Orbkit doesn’t seem to build the density from orbitals, right?
Since this seems to be getting search traffic, there’s a new pull request which will go into nightly builds as well as the next 1.99 release. Avogadro will now automatically calculate the density matrix as needed (e.g., in this case).