I think crest should be handled with some Python plugins (e.g., conda install -c conda-forge crest because there are an increasing number of features, e.g.:
Linux only (i.e., require xtb-iff):
I’m making the list partly to remind myself that it would help to add scripts for each of these types of calculations. Unfortunately the xtb-iff program needed for QCG / solvation is only available on Linux x86.
Oh, I forgot to mention that for the solvation example, geometries of the solvent are also useful, e.g.:
- water
- acetonitrile
… etc.
https://xtb-docs.readthedocs.io/en/latest/gbsa.html#parameterized-solvents
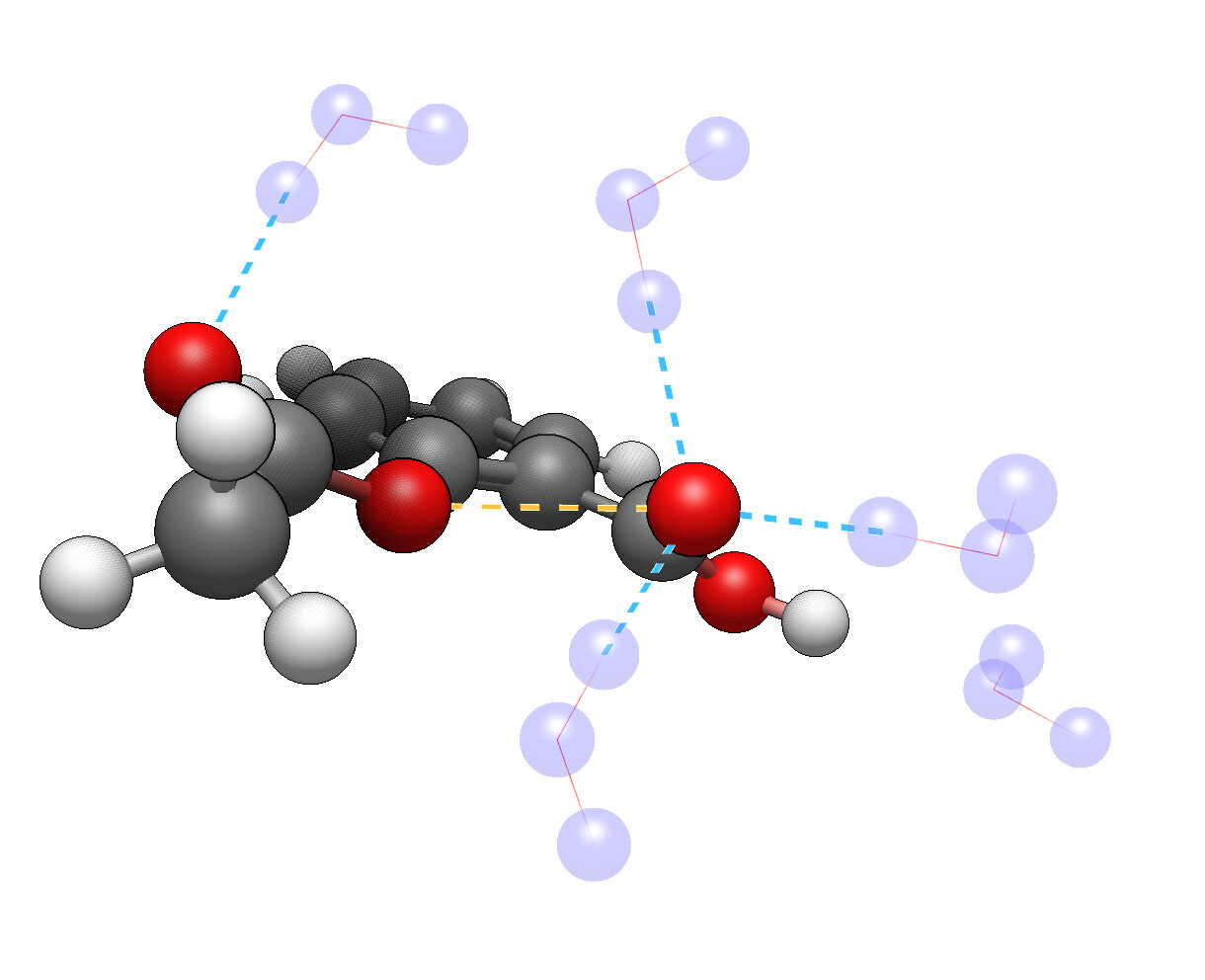
Seems like there’s a way to handle the “growing solvation” by adding solvent molecules some distance from the molecule, optimizing, then using --nci mode.
For example, here’s 5 water molecules around aspirin optimized using GFN-FF:
I’ve been thinking about somethings that I would find useful in a molecular viewing program (i.e., potential features for Avogadro 2) and they would fit it with Crest, so I thought I’d mention them here.
It would be neat to have the ability to calculate the RMSD of geometry between two conformers (or possibly more general).
It would also be great to be able to align two (or more) conformers/molecules and overlay them to visually see the difference.
I’m not quite sure how this would work, but perhaps something using the “Molecules” palette in the bottom left?
Another one (sorry). Xtb can calculate Fukui indices (for predicting reactivity). Would be useful to show this, particularly in the condensed form, so that we can visualise where reactions are likely to take place.
Fukui indices from xtb (and others) are definitely on the way. One of my students is finishing a micro-pKa model and not surprisingly, Fukui indices are useful components of the prediction.
Calculating RMSD between conformers won’t be hard. Probably something to add to a table / graph of conformers.
As far as visualizing the alignment, it’s a useful aspect for the “layers,” since you could copy / paste one conformer or molecule but into a new layer. The main new feature would be aligning the substructures.
I asked about the xib-iff program:
If you want to run QCG on Windows or Mac, I would recommend the latest build of crest by compiling it from the current source code. There, xtbiff is not required anymore as it is replaced by the aISS implemented in xtb version 6.6.0 and above.
So it sounds like the next release of crest will use the latest release of xtb and QCG for solvation will be back. (Makes sense, since the nci mode worked fairly well.)
I am getting close to a fairly finalized version of an xtb plugin. Initially it will have the following built-in calculation types:
- Single-point energy – energies returned in four units
- Optimization
- Frequency calculation – including warning for negative freqs.
- Opt + Freq – with automatic adjustment and reoptimization in case of negative frequencies
- Orbitals
- Conformer ensemble – via
crest, options specifiable
It will further have the following features:
- Menu option to open folder containing results of last calculation
- Menu option to open the xtb docs
- Configuration menu with options for solvation and parallel processing variables (cores, memory)
- A
Run... dialog allowing for:
- any valid
xtb command at all (though only certain result times will be parsed by the plugin/Avogadro, the rest will just be saved in output files)
- specification of a save location
- use of Turbomole coordinate files for periodic systems
Are there any other calculation types or features that it would be important to have for an initial release? Anyone have any ideas/requests?
Conformer ensembles will need crest but I don’t plan to make any other functionality from crest available yet. I suspect it would be better to have separate avo-xtb and avo-crest plugins, and have the other, more specialized crest run types handled by the specific plugin as proposed by @ghutchis above – agreed? Or is it more sensible to unite both? The Grimme group certainly seems to think they are best kept separate, so I’d be inclined to agree.
Yes, I think I’d separate crest into a separate plugin.
About the only thing I’d add to the current XTB list is for MD trajectories (eg -omd) since setting up the temperature, time, etc would be useful… and I’d love to use it for teaching. 
BTW, when you feel it’s ready for more widespread testing / feedback, please create a new thread and submit a pull request to GitHub - Avogadro/plugins: Scripts and data for the Avogadro plugin system
I’ve been busy with a proposal this week, but hope to finish the “install requirements.txt” components by the end of the week.
Will do! I want to implement some error catching first so that at least errors from xtb get shown to the user.
Other than error catching and calculations in parallel, I think all the basic functionality is pretty much ready. I’ve opened a pull request for the plugins repo, I just added the avo_xtb repo to it, let me know if there needs to be anything else.
Would you mind giving it a spin yourself and testing stuff before I open a thread? 
I tried the “Go to Results” and got:
FileNotFoundError: [Errno 2] No such file or directory: '/home/matt/.local/share/OpenChemistry/Avogadro/commands/avo_xtb/last'\n
This seems to be happening with a lot of the scripts now.
Ah yes, I forgot to stop tracking config.json with git. So it hasn’t generated a new config file as it should but is instead reading my config and throwing the error because obviously it is not finding things in the same place as on my machine. If you delete config.json from the plugin’s directory then it should all work?
Yes, looks like that solves the problem.