Wanting to make Bisphenol-A. Inserting a phenol ‘fragment’ is fine, but after adding the second, both are coupled for all movements. Need to rotate one of them then mix in acetone then rebond.
Unfocus does not seem to work.
Please advise.
Wanting to make Bisphenol-A. Inserting a phenol ‘fragment’ is fine, but after adding the second, both are coupled for all movements. Need to rotate one of them then mix in acetone then rebond.
Unfocus does not seem to work.
Please advise.
I’m guessing here, but assume that both fragments are selected (e.g., the light transparent blue spheres around the atoms)?
After inserting the first fragment, try Select => Select None (or Control-Shift-A) to unselect that one before inserting the second.
The “unfocus” is in the view menu - it resets the view camera, not anything with the model.
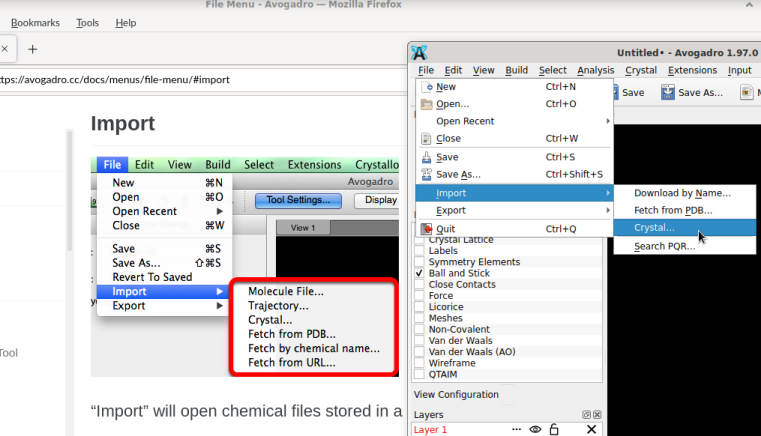
BTW, you can also build Bisphenol A through File => Import => Download By Name or by grabbing the SMILES from Wikipedia: CC(C)(c1ccc(cc1)O)c2ccc(cc2)O
Hope that helps!
Thanks, it’s working. Trying to work on a project with lots of molecules to create and optimize etc.
More items (if you like I can repost on forum):
@ghutchis Would contacting the cactus server at NIH by Avogadro an interesting feature to add as a potentially larger public data repository about molecules than Wikipedia?
Because when running Jmol* on a computer connected to the internet, this allows to resolve (trade) names, CAS registry numbers, SMILES, etc. with their public database. Frequently, this is faster than to plug and join atoms and bonds manually, just from the program’s console (file → console):
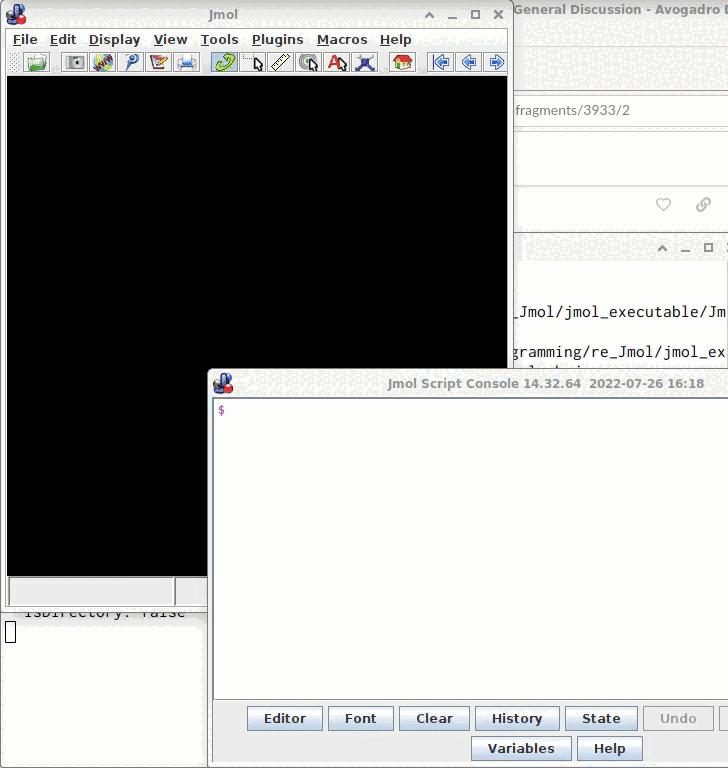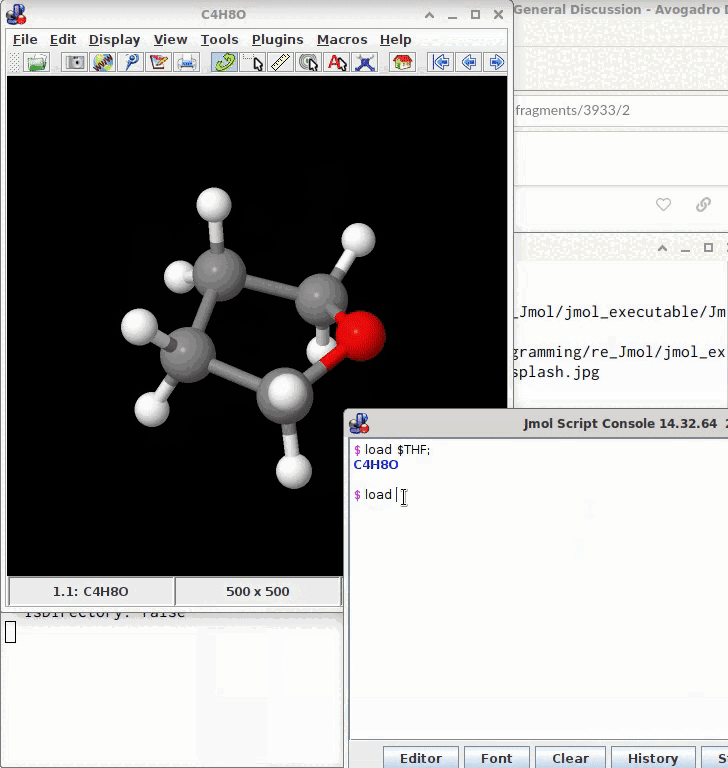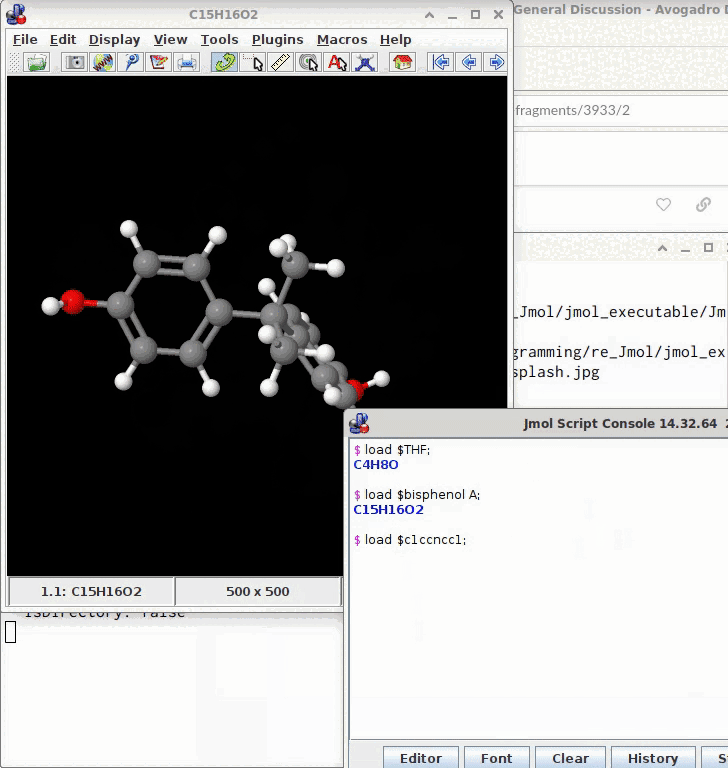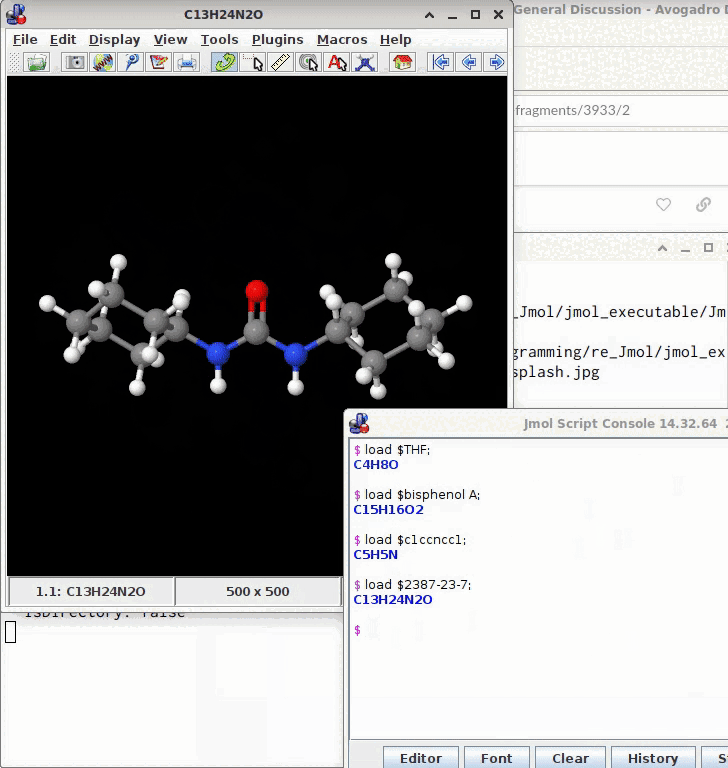
@timh The model may be saved from the console by a subsequent write model.sdf; (or as a .mol, .xyz; .png etc.)
*) For this, the program doesn’t need to nest into the operating system. If there already is Java to run the program /at all/, this equally works out-of-the box right after unzipping the archive from sourceforge.
Yes, this feature has existed in every version of Avogadro. I mentioned it above. It’s:
File => Import => Download by Name
I mentioned inserting by SMILES because sometimes a molecule isn’t in the resolver, but you have the SMILES.
Thank you for the clarification. It shows I misunderstood your mention of wikipedia as accessing the small (about 20k entries) set as available e.g., via cheminfo.org. Because the source isn’t mentioned in the manual when describing the file menu, but when describing the molecule’s import by name.
Along this way, I noticed the current version of the manual and the GUI (as for 1.97.0, app image of Linux) occasionally differ about each other, e.g. the two addresses (revision stamp May 23, 2022) and permitted imports
and the assignment of names now moved to analysis → properties → molecular, yet yielding a reorganized widget currently missing the very line reporting the anticipated property.
This is kinda off-thread, but yes, the manual is written for Avogadro 1.2, and the app is not. (I only have 24 hours in a day, sadly.)
As far as the name assignment, that’s been merged into the development code, e.g.
I think you mean they’ll have problems in GAMESS, etc. If you do “Optimize Geometry” in Avogadro, it will use a force field to clean up the structure.
Quantum chemical programs won’t like long bonds … if you stretch a bond, is that homolytic cleavage? Heterolytic? Some superposition of both?
I’m not involved with MoleQueue. I’d like to see it work, but at the moment it’s somewhat abandoned. The main thing is to set up “how to run” programs, either locally or on a queue - more documentation and example scripts than anything else.
Great! If PPG is interested, I’d be more than happy to come out and give a workshop and/or look at particular problems / bugs.