Avogadro version: 1.100.0-7-gac3f38f (Nightly from today)
Operating system and version: Windows 11 Pro 24H2
Expected Behavior
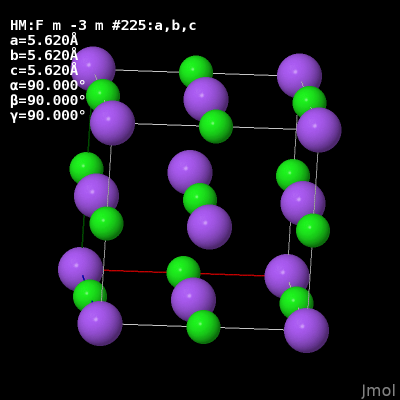
Using Crystal>Fill Unit Cell command should fill all atoms in a unit cell.
Actual Behavior
The cell is not completely filled, atoms on the edge do not seem to be filled. For example, in a BCC unit cell there should be atoms at all eight corners and one in the center, however only the original atom at the origin and the atom in the center are present after using the command.
Steps to Reproduce
Import a crystal structure (I used Mo, V, Li, NaCl, and AlAs) and use the Fill Unit Cell command.
Also, as a side note, I noticed that there is no crystal structure in the Elements set for Polonium.
They’re not because those positions are translationally equivalent.
The previous versions would generate everything. The problem is that if you go to run a calculation, you’ll get errors because you have multiple atoms in translationally equivalent positions.
I understand for some visualization purposes it’s nice to have “all” the positions. For 1.100, I didn’t want to add a new command, since that would require a new string for translation.
I’m willing to take suggestions for the wording of “Fill Unit Cell” with all positions. The code is there, I just don’t know a good way to word it. (And honestly, I think it needs some sort of warning.)
Crystal structures Avogadro2 provides are derived from the public COD. If you search for elemental polonium, this database has three records for the element, COD 1010519 by ED in 1936; as well as two different XRD based models by 1949, COD 1509138 and COD 1537759.
If you query the ISCD about inorganic structure data, additional indexed literature data about the element are (though not always with coordinates deposited):
ISCD 43211 and ISCD 43212 about a publication by R.J. Desando, R.C. Lange, Journal of Inorganic and Nuclear Chemistry, 1966, 28, 1837 (DOI 10.1016/0022-1902(66)80270-1)
ICSD 1887509 by A. Rubio-Ponce, J. Morales, D. Olguin, Progress in Theoretical Chemistry and Physics, 2012, 22, 119, DOI: 10.1007/978-94-007-2076-3_7
ICSD 426966 by Kurt Lejaeghere, Veronique Van Speybroeck, Guido Van Oost, Stefaan Cottenier, Critical Reviews in Solid State and Materials Sciences, 2014, 39, 1, DOI: 10.1080/10408436.2013.772503
ICSD 649161 by W.H. Beamer, C.R. Maxwell, AIP The Journal of Chemical Physics, 1949, 17, 1293, DOI: 10.1063/1.1747155
ICSD 649163 by R.J. Desando, R.C. Lange, Journal of Inorganic and Nuclear Chemistry, 1966, 28, 1837, DOI: 10.1016/0022-1902(66)80270-1
ISCD 655031 by W.H. Beamer, C.R. Maxwell, AIP The Journal of Chemical Physics, 1949, 17, 1293, DOI: 10.1063/1.1747155
ISCD (like the CSD file by CCDC more focused on small organic structures) is restrictive regarding use and share of the structure data. The COD on the other hand not only is permissive, it encourages further share right on its landing page with “All data on this site have been placed in the public domain by the contributors.” – its data and tools alike. Hence the choice of Avogadro2 to use COD data. However, given the time the three COD models were established, and the then available technology (wet-chemistry films running Weissenberg- and Buerger precession cameras as shown here) it is much more difficult to obtain a good data set (and eventually, a good crystallographic model) than with modern diffractometers and their (computerized) data processing. This likely contributed to leave them out, aside of Po and its radioactivity …
No, omitting Po was simply an oversight. Not quite sure how it happened…
If someone wants to select a designated entry from COD, I’m happy to update the repository. I’ll point out that it has the 1949 Beamer paper (both the cubic unit cell and the hexagonal).
It looks like if you include theoretical results, COD also has the Critical Reviews paper from 2014 as 1512532. That’s probably my preference.
Regarding theoretical results, CCDC Cambridge/UK is one organizer of blind tests where participants are invited to speculate for crystal structures about records of the CSD file which are not yet accessible by the regular users of the database. Most recent is round 7 with results published in December 2024 (Acta Cryst. (2024). B80, 517–547, DOI 10.1107/S2052520624007492, open access). Since tests like these are interesting for method development, the stewards of the COD equally set up TCOD* as a dedicated “Open-access collection of theoretically calculated or refined crystal structures of organic, inorganic, metal-organic compounds and minerals, excluding biopolymers.” Thus it not too surprising record COD 1512532 mirrors the record TCOD 20000147.
* And with similar infrastructure, they built one about Raman spectroscopy, the ROD, too.
I think it’s good, although people may be confused about whether or not it will fill the entire unit cell or only the atoms on the edges.
Perhaps changing the original from “Fill Unit Cell” to “Fill Unique Positions” and keeping the “Fill Unit Cell” the same, as that was the original behavior of the command. Something like that may avoid any confusion about the function of each command?
We could change “Fill Unit Cell” to “Complete Unit Cell”? The current behaviour is the correct one though, so please let’s not change it.
I actually think a command to do what @brockdyer03 is looking for, as in, duplicate atoms on edges, would not be a great approach. But if you wanted to have a command like that, a name could be “Duplicate Atoms on Faces” maybe? I don’t think “Fill” is an appropriate term because it implies they are supposed to be there.
Ideally instead there would be some purely visual tricks to help with visualization that achieve the same thing but don’t actually change the atom set, thus maintaining accuracy.
I came up with a couple of nice options, but this one seems the best to me:
How about the spheres of atoms wrap at the unit cell boundary?
I.e. they are shown as hemispheres or quarters or whatever fraction is appropriate (the cell boundary slices them). The atom chunks on each face are all the same atom, so moving one causes all the others to move. Moving it off of 0 doesn’t cause the other parts to vanish, the section of the sphere that crosses the cell boundary continues to wrap appropriately, and bringing any atom near to the boundary causes the wrapping to kick in.
Wouldn’t this be crazy nice if it were possible? Imagine the educational value of being able to click and drag a face atom and get the visual feedback that the one on the opposite face is the “same” atom…
To begin with I thought it’d be very hard to code, but actually, if all coordinates – universally – in a unit cell environment were a different class to the normal ones and that class had built-in overflow at 1.0, then I feel like the behaviour would kind of automatically follow? Maybe that’s a naive understanding.
I reckon what we’d also get for free is that bonds would then wrap too and moving one of the atoms would cause both “halves” of the bond to move appropriately.
Right now, atom spheres are drawn using “impostors.” Basically, you write a shader program that tells the GPU to render a perfect sphere at a particular point. Then you give a bunch of points and the GPU takes care of the work.
In your case, with bonds or atoms getting sliced by the unit cell … you have to compute all those intersections and either have a very flexible sphere rendering code, or a set of pre-sliced spheres / cylinders. (Maybe there’s a better way, I’m certainly not a shader expert.)
I certainly agree with the intuition / educational perspective, and it’s certainly something to work towards (we had sliced bonds in some Avogadro 1.2 builds). But it’s a hard thing to implement.
That seems like a good wording. I don’t think I’m going to change “Fill Unit Cell” because it’s something that’s been used for a while in Avogadro and Open Babel.
This might be a bit easier (e.g., rendering the translationally equivalent atoms, while keeping the symmetry unique versions). In this case, for example the VdW spheres code would have an option to show the translationally equivalent atoms - and if there’s a unit cell in the molecule, you’d generate the translational copies when rendering to the screen.
@matterhorn103 Briefly I thought your line of argumentation were to offer a supplementary style of representation of a unit cell, i.e. not only the one with “full spheres”
regardless if they are within the parallelepiped and count 1/1, on a face (1/2), ridge/arête (1/4), or apex (1/8), but capped
If one of the atoms “happens” to be at (0,0,0) either a) because the asymmetric unit defines them at this spot, or b) by filling the unit cell (i.e., asymmetric unit + application of the symmetry operators of the unit cell) I presumed the origin of the coordinate about the enveloping “shoe box” (where \vec{a}, \vec{b}, \vec{c} in the display of CCDC’s Mercury, or here Jmol “emerge”) would automatically recenter and realign.
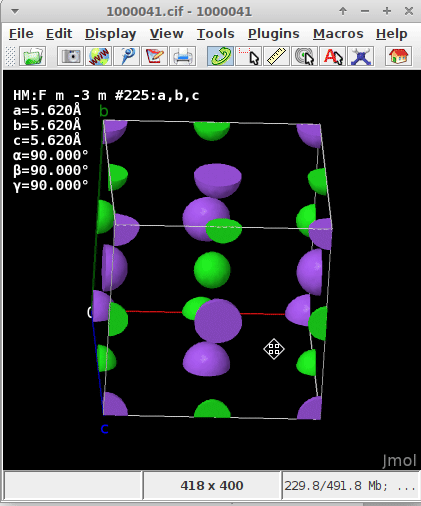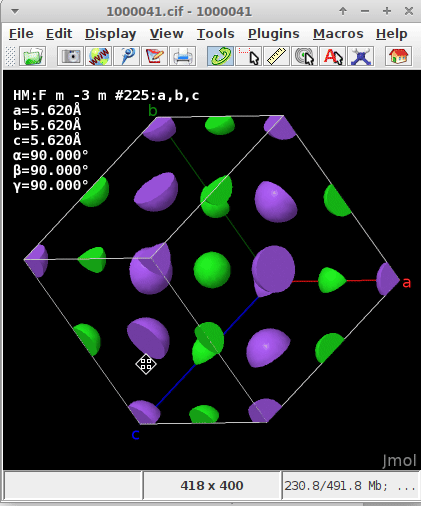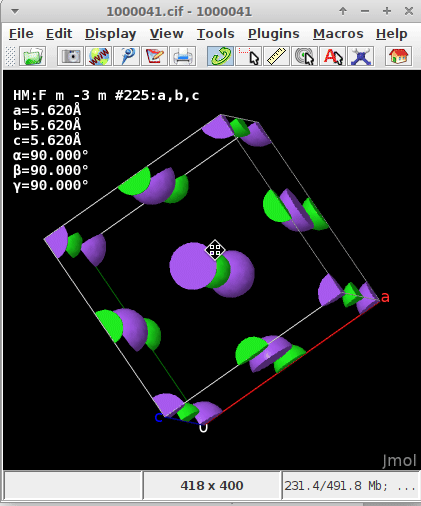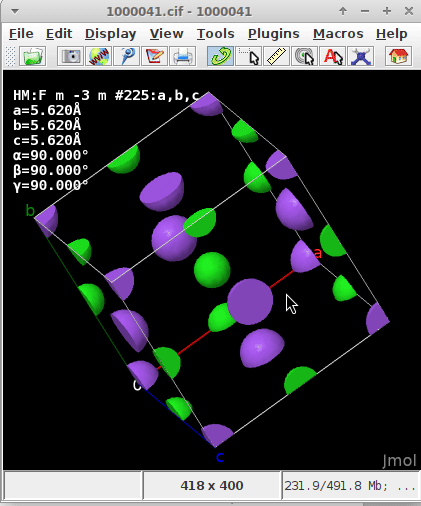
The .cif read for the static/dynamic visualization is plain NaCl COD 1000041.
The commands issued to Jmol after downloading the .cif were