As I mentioned in the June Development Update, I’ve integrated a new framework for charges / electrostatics. It’s intended to be fairly general, since there are a variety of models to evaluate molecular electrostatics, not just sets of atomic partial charges.
I posted on Twitter, but I’m also interested in getting feedback on here. You’ll be able to write Python scripts to get atomic partial charges and/or electrostatic potential at sets of points (e.g., a molecular surface).
Here’s what I’ve heard so far:
What else? Is there an easy way to assign AM1-BCC charges?
Any other packages you use to assign partial charges or electrostatics?
But here’s another question …
Should we scale red/blue to the charges in the molecule or to an absolute scale (e.g., -1 to +1)?
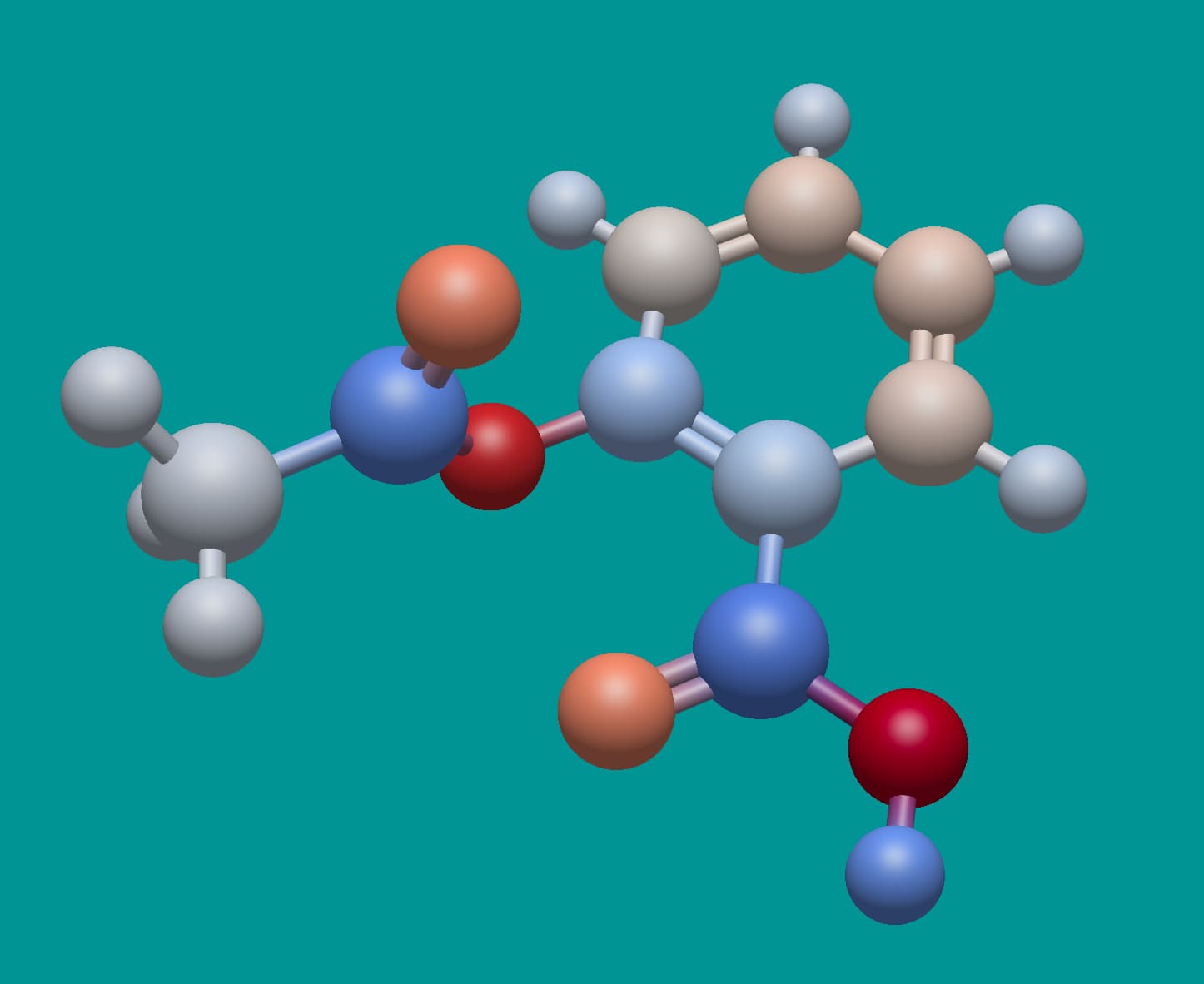
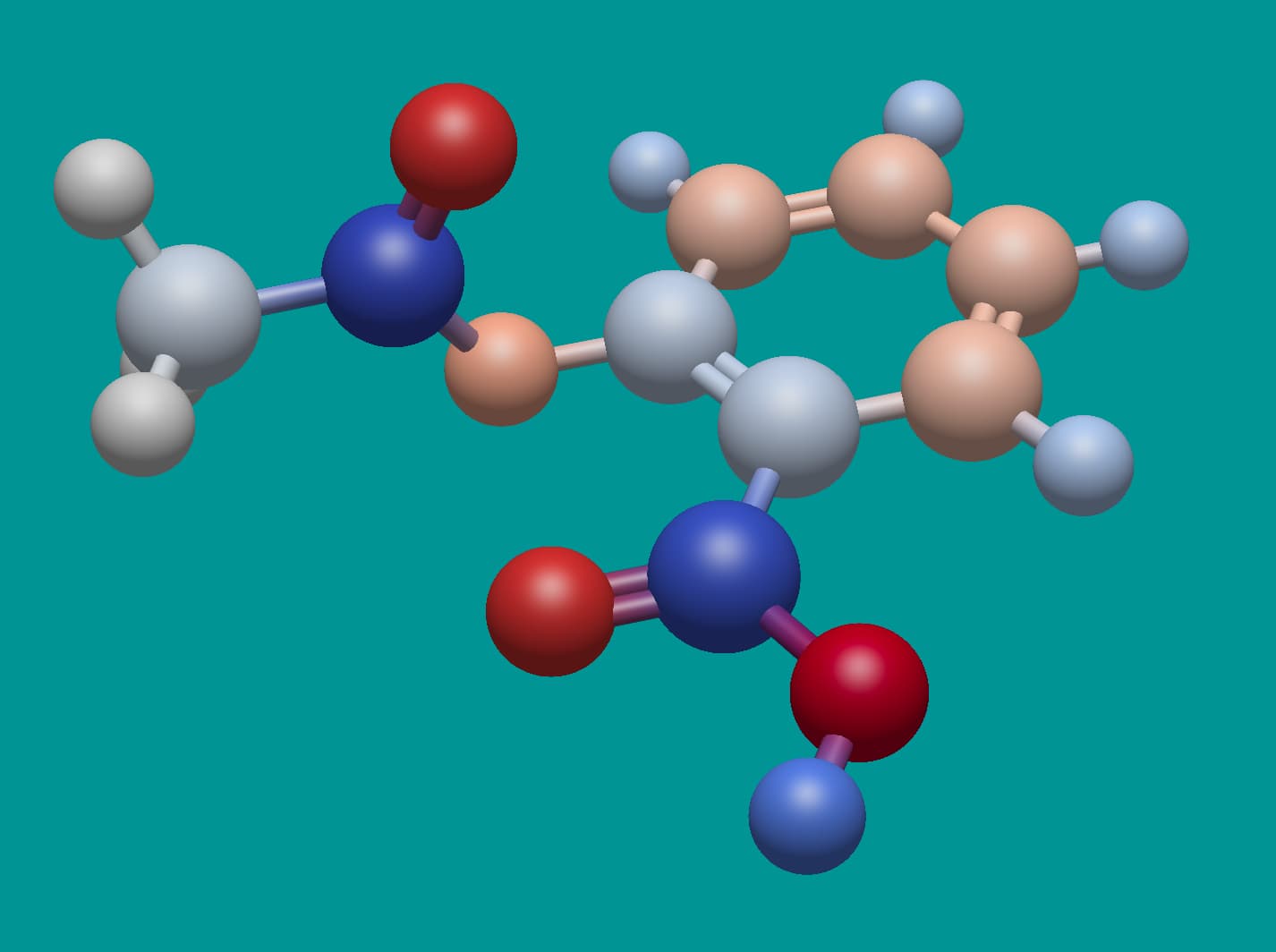
Here’s aspirin. Notice the deep red … those oxygen atoms have a charge of -0.48. Should they be so strong?
I’m leaning towards yes, because these empirical charge models are all relative.
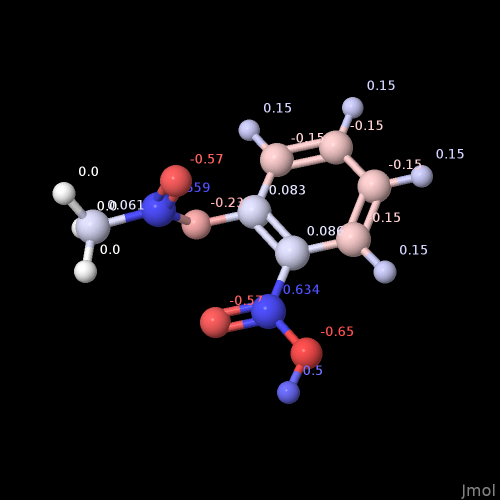
Right of the tip of my hat I do not recall which model is used in Jmol to assign the partial charges, but (perhaps with exception of O along C-O-C of the ester) the pattern looks very similar to the one depicted by you:
(prepared with Jmol version 14.32.60, released by 2022-06-06).
To replicate either:
-
via GUI: 1) load a structure .sdf with aspirine, 2) right mouse click on the canvas of the program to open the optional pull-down menu (not the one attached to the frame), select color → by scheme → partial charge.
-
Or 2), launch Jmol, then run File → Console for its own terminal. The sequence of commands used to generate the .png was:
load $aspirine; # the sole step relying on internet contact to NIH/cactus
color partialcharge;
label %[partialcharge];
set labeloffset 10 10;
# now reorient the model with the mouse
write "aspirine_Jmol.png";
exitJmol;
As far as I can tell, that’s from the MMFF94 partial charge scheme.
Do you know or remember if Jmol scales the colormap based on the charges (e.g., -0.65 / -0.57) or sticks to an absolute scale?
(And it’s a good reminder that I need to add partial charge labels for atoms.)
This has been merged, and includes example scripts for GFN2 and AM1-BCC charge models.
Affirmative for Jmol and using MMFF94. The assignment and subsequent of partial charges entered Jmol with version 12.3.24 distributed by May 2012 (entry 1, entry 2).
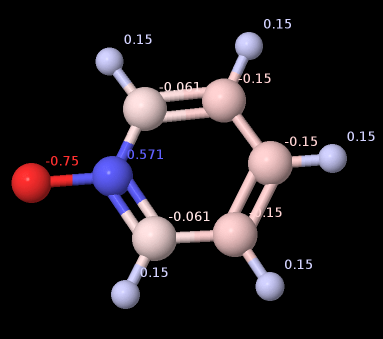
While atoms of “normal organic molecules” remain in the interval of [-1, +1], including e.g. N-pyridine oxide
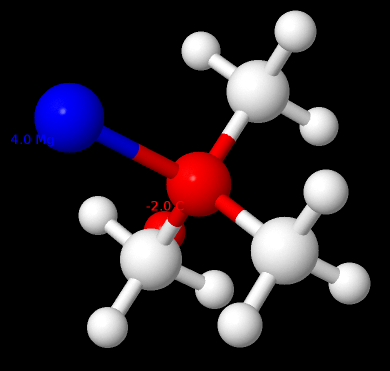
I just noticed Jmol can display decimals outside this range. Only as example this is possible by the numbers, tert-butyl magnesium chloride displays -2.0 on the tertiary C and + 4.0 Mg:
with current Jmol (14.32.62) and loading the data on Jmol’s console by either
load $pyridine-N-oxide;
load $$tertbutyl-magnesiumchloride;
But I assume the underlying theory and model support only in case of the N-oxide the .sdf amended by the block of partial charges:
https://cactus.nci.nih.gov/chemical/structure/pyridine-N-oxide/file?format=sd...
__Jmol-14_07062200000D 1 1.00000 0.00000 0
Jmol version 14.32.62 2022-06-29 00:00 EXTRACT: ({0:11})
12 12 0 0 0 0 999 V2000
-0.8768 -0.0000 0.0020 N 0 3 0 0 0 0
-0.2338 -1.1527 0.0010 C 0 0 0 0 0 0
1.1474 -1.1913 -0.0003 C 0 0 0 0 0 0
1.8569 0.0000 -0.0011 C 0 0 0 0 0 0
1.1474 1.1913 -0.0005 C 0 0 0 0 0 0
-0.2338 1.1527 0.0013 C 0 0 0 0 0 0
-2.2928 -0.0000 -0.0021 O 0 5 0 0 0 0
-0.7937 -2.0762 0.0017 H 0 0 0 0 0 0
1.6679 -2.1376 -0.0004 H 0 0 0 0 0 0
2.9369 0.0000 -0.0018 H 0 0 0 0 0 0
1.6679 2.1376 -0.0007 H 0 0 0 0 0 0
-0.7937 2.0762 0.0014 H 0 0 0 0 0 0
1 2 2 0 0 0
2 3 1 0 0 0
3 4 2 0 0 0
4 5 1 0 0 0
5 6 2 0 0 0
1 6 1 0 0 0
1 7 1 0 0 0
2 8 1 0 0 0
3 9 1 0 0 0
4 10 1 0 0 0
5 11 1 0 0 0
6 12 1 0 0 0
M END
> <JMOL_ATOM_NAMES>
12
1 N1
2 C2
3 C3
4 C4
5 C5
6 C6
7 O7
8 H8
9 H9
10 H10
11 H11
12 H12
> <JMOL_PARTIAL_CHARGES>
12
1 0.571
2 -0.061
3 -0.15
4 -0.15
5 -0.15
6 -0.061
7 -0.75
8 0.15
9 0.15
10 0.15
11 0.15
12 0.15
$$$$