I tried to download Avogadro 1 for my college Orgo class. As a part of this class, we use Avogadro 1 to build models of compounds, and calculate the in-space distance, dihedral angles, and total energy of various molecules and their conformers.
I am using a Lenovo Ideapad flex computer running on Windows 11 version 22H2. When I download Avogadro 1 version 1.2, I am unable to build molecules because I cannot build carbon chains. Here is my specific issue:
when I click with the draw tool, it generates 1 methane, but when I try to click on one of the connected hydrogens to turn it into a carbon and build a carbon chain, nothing happens, or it makes another methane that IS NOT bonded to the original carbon. I also cannot drag between two carbons to create a bond between them. Nothing happens. This is very problematic for my class. None of my classmates have had the same issue. I have also restarted my computer countless times and reinstalled the software multiple times. Can someone tell me how to fix this issue? The only way that I can generate a carbon chain is by inserting it with Insert → fragment.
When I use avogadro 2 (version 1.97), I do not have these issues. I am able to build molecules without any problem. The issue is that I don’t know how to do the “calculate energy” function on this version of the software. Avogadro 1 has an extension for molecular mechanics to “calculate energy,” but I don’t see the same option for Avogadro 2. I am graded on the values that I calculate for between the space distance and energy of compounds, so I need these values to exactly match those attained by the version of Avogadro 1 that my classmates and teacher are using. Is there a way to get the molecular mechanics function downloaded on Avogadro version 1.97? Am I missing something?
I am happy to provide more information if anyone needs. please help me soon.
Does the coursework tell you which molecular mechanics model you need to use? If I remember correctly, Avogadro 1 uses Open Babel’s forcefield by default (I am sure I’ll be corrected if I’m wrong) in which case the functionality is still available in Avo 2.
I don’t have a copy of Avogadro to hand right now, but isn’t there a “Calculate Energy” option under Extensions > Open Babel?
My professor just tells us to use the one that avogadro 1 automatically uses. I’m not sure which extension it is. I think that he told us to use the MMFF94 force field though, and leave everything else the same
When I go to the Open Babel section of the extensions tab on Avogadro 2, I do not see an option to calculate energy. I see the following options:
add hydrogens
add hydrogens for pH
Configure force field
Optimize geometry
perceive bonds
remove hydrogens
Am I missing something? I have tried to look at all of the tabs and even other tabs as well. I tried to select the molecule and do the configure force field option, but nothing happened. I don’t even know what that tool does/what is supposed to happen.
When using Avogadro 1, it seems that this energy calculating function is built in. Is there a way that I can download another extension to avogadro 2 that uses the same mechanics as Avogadro 1 to calculate energy?
Avogadro 1.98.1 has “Calculate Energy” under the Open Babel section. I didn’t realize it wasn’t available before 1.98.x (I don’t use force field energies.)
Alternatively, you can build the molecule you want, save it as CML and open in Avogadro 1.2.
Thank you for the feedback! Unfortunately, I still encountered some issues. I downloaded Avogadro 1.98.1,
To start, the energy calculations were in LJ, rather than KJ/Mol and the values did not match up with what my friends found using their version of Avogadro. It seems that the calculate > energy function is not a part of the open babel section, rather its own section titles “calculate”. Is there a way to convert from LJ to the desired value?
The energy of an optomize CH4 was 6.509 e+07, and my classmates found that it was 0.1105 kJ/mol (desired)
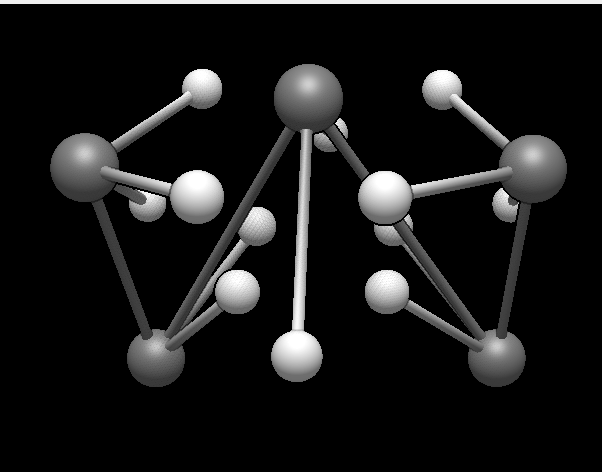
Under this same calculate tab is an “optimize” button that creates very very weird conformers. This was a simple pentane that was optimized by the software. It looks really weird. Does anyone know what is going on with this?
Fortunately, the workaround that you mentioned where I build the molecule in Avogadro 2 and upload it into Avogadro 1 does work! although it is a little bit clunky. Thank you.
Does anyone have any other recommendations by chance?
I tried using the optimization feature of Avogadro 1.98.1 on cholesterol, then importing that optimized CML file into avogadro 1, but it seems that the optimization created a molecule with different energies in avogadro 2 than avogadro 1. Although the molecules looked similar they had vastly different energy calculations when uploaded into avogadro 1. the avogadro 2 was 2.6 e+11, while the avogadro 1 was 397.033 (kj/mol).
Additionally when I was using avogadro 2, it would crash often (just closing completely), or not optimize, instead saying “unable to open open babel optimization”
From this investigation, it seems that avogadro 2’s optimization is different than that of avogadro 1. Does anyone know how to solve my origional problem with avogadro 1, not being able to add in new carbons?
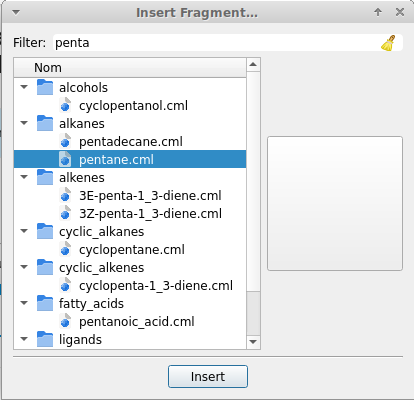
@drepang04 For a structure this simple as n-pentane you don’t need to build the molecule by yourself.
pentane is considered a motif important enough that Build → Insert → Fragment offers you to add to the current scene the complete molecule in an already optimized geometry. Either check the section alkanes, or type the chemical name to retrieve it:
As you see, once you entered enough characters, Avogadro is smart enough to provide you suggestions to choose from.
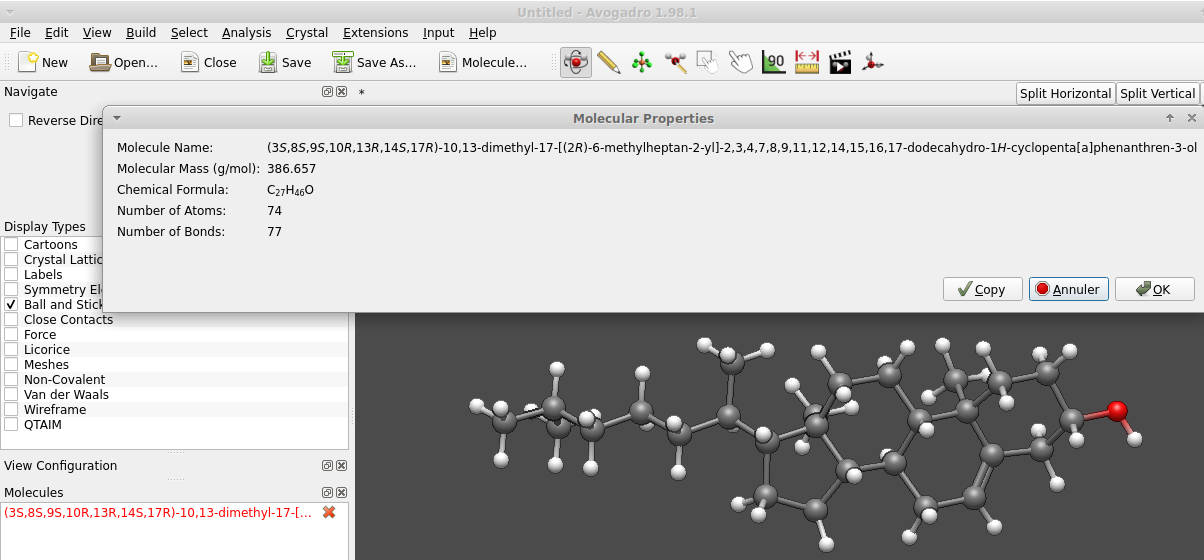
In case a molecule (or a suitable skeleton of a molecule to edit manually) is not in the library of fragments, one reasonable conformer can be imported (File → Import → Download by Name, then type the chemical name) from servers of the NIH (I think). This again much faster than to sketch atoms and bond one by one. Even more so as the mere input of cholesterol instead of the systematic name rapidly provides the 74 atoms joined by 77 bonds:
I guess you are using Extensions > Calculate > Optimize. This uses the new molecular mechanics framework and can use many different forcefields, but by default (without adding extra ones yourself) only the Lennard-Jones potential is available, which gives the results that you are seeing.
Isn’t there an option at Extensions > Open Babel > Optimize, which should give you what you want? Alternatively the shortcut Ctrl+Alt+O for an optimization and Ctrl+Alt+E for an energy should also invoke that Open Babel forcefield, which I assume is still the same forcefield as in Avogadro 1 and should then give the same results.