Hello, I’ve run into a problem with optimization that occurs in this error code:
“Cannot set up the currently selected force field for this molecule. Switching to UFF.”
At first I thought it was because I’m working with a large molecule & 2 others but that doesn’t seem to be the case, only when I flipped the carbon chain around did this happen… is there any way I can flip the carbon chain & still have the force field use MMFF94?
I think I’d need to see your molecule / file. It shouldn’t have anything by to do with the geometry, only elements and in some cases charge / etc.
Oay!
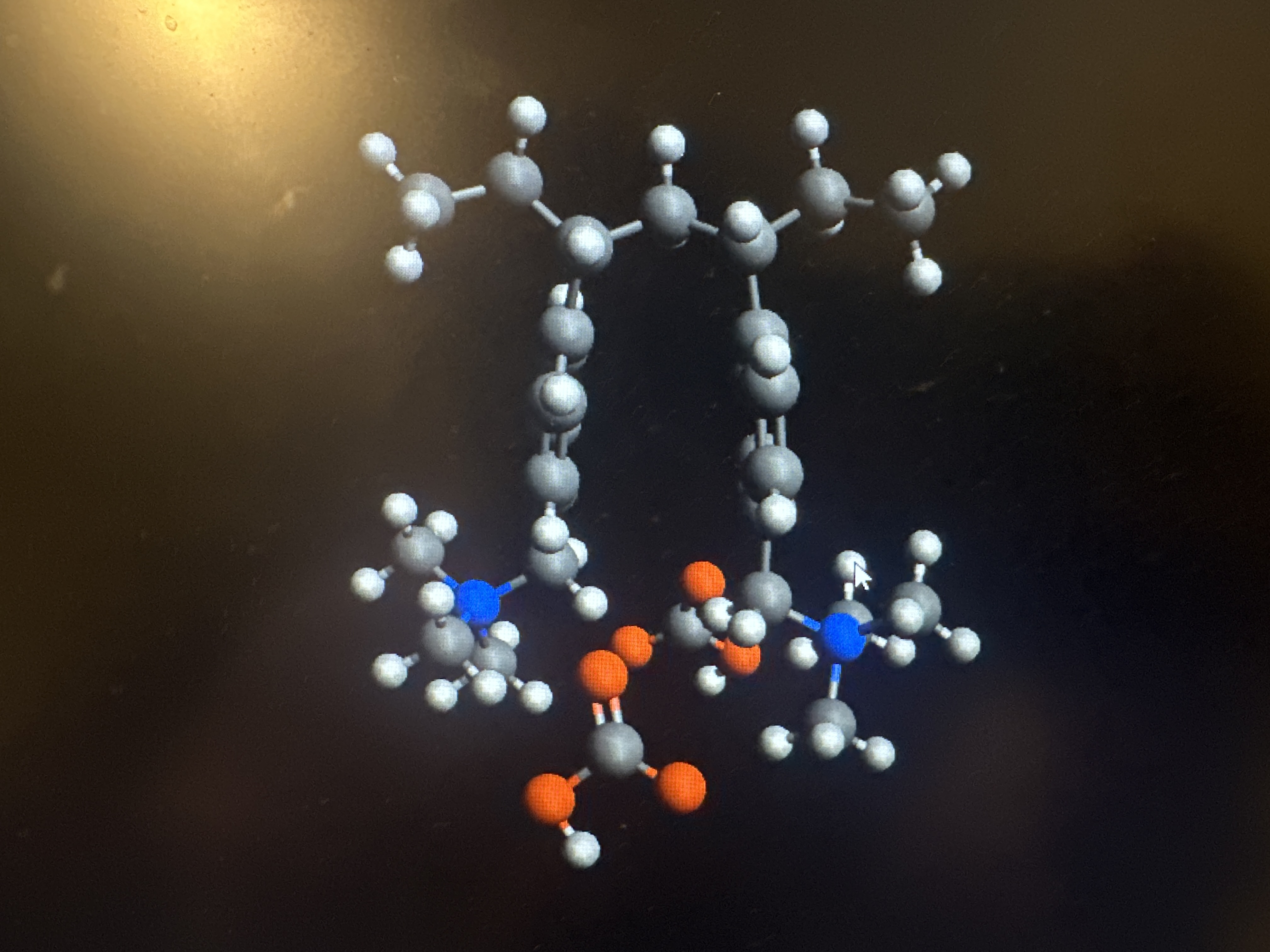
3 Carbon Gap Dry
This is the file that does not work vs the one I had changed it from:
I’m not sure exactly how it happened (only that I’d suggest saving in .mol / .sdf instead of .pdb) but you can see the difference if you look at the text of the files:
2 Carbon Gap
HETATM 1 C UNK 0 -3.013 2.635 0.048 1.00 0.00 C
HETATM 2 C UNK 0 -3.618 3.632 -0.935 1.00 0.00 C
HETATM 3 H UNK 0 -3.569 1.693 0.015 1.00 0.00 H
HETATM 4 H UNK 0 -1.969 2.412 -0.194 1.00 0.00 H
HETATM 5 H UNK 0 -3.057 3.018 1.072 1.00 0.00 H
HETATM 6 C UNK 0 -2.968 5.035 -0.863 1.00 0.00 C
HETATM 7 H UNK 0 -3.553 3.221 -1.950 1.00 0.00 H
Note the elements in the column on the right. All neutral atoms.
3 Carbon Gap:
HETATM 1 C UNK 0 -6.257 9.594 0.854 1.00 0.00 C
HETATM 2 C UNK 0 -5.434 8.632 0.004 1.00 0.00 C
HETATM 3 H UNK 0 -5.760 10.567 0.906 1.00 0.00 H
HETATM 4 H UNK 0 -7.255 9.754 0.432 1.00 0.00 H
HETATM 5 H UNK 0 -6.370 9.217 1.875 1.00 0.00 H
HETATM 6 C UNK 0 -6.005 7.193 -0.020 1.00 0.00 C1-
Note that several of the atoms include charges, e.g. C1- which would be a carbanion. MMFF94 doesn’t have parameters for that. (Technically UFF doesn’t either, but this UFF implementation will approximate parameters as needed.)
As a workaround, I’d suggest saving these files to XYZ (which doesn’t track atom charges, bonds, etc.)
You will probably need to adjust the bonds to your liking after re-reading from XYZ … because Avogadro / Open Babel needs to re-guess the bonds and bond orders. I saw several broken bonds, etc.
But once you do that, MMFF94 should be fine … the code just saw carbon anions and went ![]() and switched to UFF as the fallback.
and switched to UFF as the fallback.