After using Avogadro for many months, and quite a long amount of time in the past few days (if there was a tracker of how many hours I spent using it, I’d be in the hundreds by now), I have some QOL suggestions for the development folks. I am very open to feedback on these, and I know for a fact most of them are just my own personal preferences so I figure there might be a few who disagree. Regardless, here are some thoughts I have:
Create some hotkeys for various menu options
a) I usually need to switch quickly between several tools, i.e. the build tool, view tool, manipulation tool, and selection tool, and having to click up at the top every time can get a bit tedious
The addition of some shortcuts to various commonly used things would be great!
a) I often need to visualize molecular orbitals, and having to work through the analysis menu can be annoying when I am working on a lot of molecules. A shortcut for generating the HOMO or LUMO would be super nice to have!
b) I am positive it would be challenging, but the ability to create custom shortcuts would be a complete game changer for my workflow with A2, and I may look into designing that myself as a plugin if I ever find time away from my research…
c) If this is too much work for what it is worth, I would also not mind having the default molecular orbital in the surfaces menu be the HOMO instead of the LUMO, just a little nitpick there though.
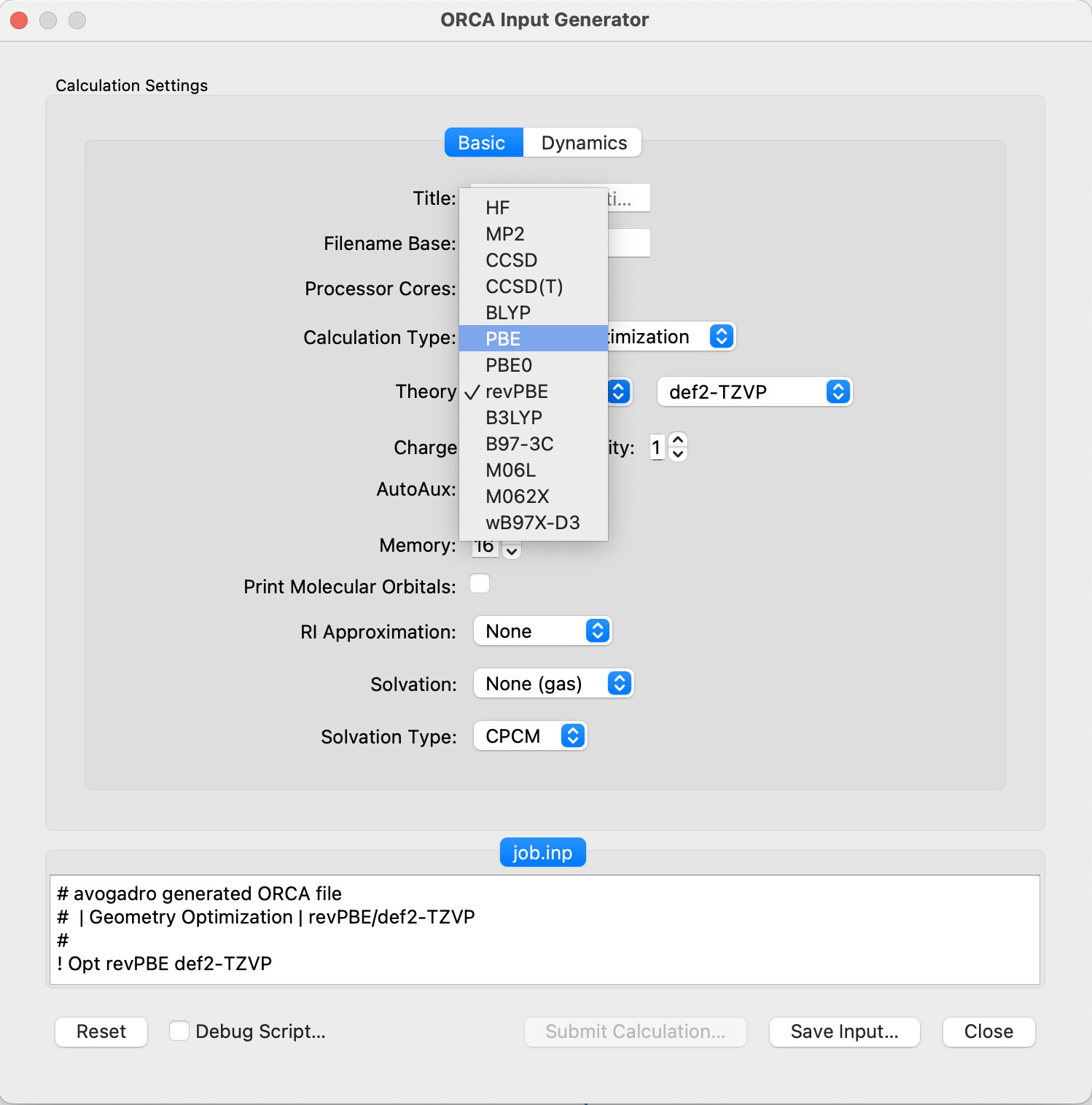
The input generators are super nice, although I find myself often needing to manually edit the inputs after A2 makes them, so a way to add a template input that can be manually set would be great
a) I use ORCA for basically every serious calculation, and my method stays the same every time, optimize with wB97X-D3BJ/ma-def2-TZVP, and then recalculate energies with DLPNO-CCSD(T)/aug-cc-pVTZ. I have a standard input block that I copy and paste into my inputs, but if A2 already had that, I would be able to save a lot of time!
Displaying molecule information when reading in from a file!
a) Very often I scroll through huge output files to find, on average, 3 numbers: single point energy, HOMO energy, and LUMO energy. I wrote a program that does some of that automatically, but often I find myself jumping between A2 and output files to look at the energies of a molecule. Having a small script that just pulls those values out of the output file and puts them in, for instance, the top left corner of the display box would be super helpful.
When I am splitting my screen (split horizontal and split vertical in the top right), my view settings aren’t retained. My projection goes from orthographic to perspective, it re-enables ambient occlusion, and the background is transparent. Having the new screen just retain the properties of the previous one would save folks a bunch of time having to fiddle around with the settings again.
That’s most of what I have been thinking recently, though I have no doubt that there are other folks with other ideas. If anyone has feedback just let me know!
On the Avogadro2 side, the main task is to expand the “molecular properties” window to include additional properties like this (e.g., into a small table).
Excellent stuff, I had no idea using ctrl+number would switch the menus! I took the liberty of updating the orca.py file in A2, made my own template that is just hard-coded into the python script.
I think that I might want to modify that script a bit further to be able to save a text file with custom extensions, e.g. a user could select an option that says “Save Template” and it would use the options they had selected to create a file that could be read over and over. Pretty sure I can get that done on my end, and if things seem to work out well I’ll put something on the github (as soon as I remember to check out the link you had given me for contributing guidelines).
I do agree with the other users that the orbital window should come back, not sure how that works but I am confident that at some point you’ll get it done, or I’ll get an itch to learn C++ and do it myself. Either way, it’ll eventually get done.
I have been writing my own script to parse ORCA outputs as a way to teach myself some more python, but it is interesting that the code needed to do that is already part of cclib, that would make implementing it into A2 much easier I feel. I think that expanding the molecular properties window might be something that I try to accomplish myself, depending on how hard it is to learn the requisite C++.
From what I can tell, it seems like all I’d have to do is check the part of the A2 libs that create that window, and then use cclib (not sure how all that works, but I am sure I will figure it out) to extract the requisite data from the output file. In my mind all I would want to include there is some stuff like the final single point energy, the HOMO and LUMO energies, and perhaps implement something that could search for thermodynamic values and pull those.
I’ll be checking that out soon, but for now I get to turn my old desktop into a server that I can use to run ORCA calculations remotely. Yay Linux!
There’s a bit of Qt to create a scrollable table, but I’ll create a branch … and then you can feel free to add some more properties.
At the moment, there’s a cclib plugin for Avogadro2 but also a separate C++ implementation. So there may need to be some help here eventually (e.g., to get all the properties you might want):
Yes, this has been on my mind for a while. The “Save Template” is pretty easy. The question on my mind is how the UI works once you save the template. Does the Input Generator dialog default to the template? Does it ignore the template once you start changing options? etc.
Basically, if we can figure out how the feature should work, it won’t be too hard to do.
One solution might be the ability to add/customise menu options.
Programs like Orca and Gaussian use keywords that are placed in a simple input line to specify method, basis set etc. Avogadro 2 allows you to choose between a selection of common methods through the dropdown menu:
One solution would be to have an extra option, something like ‘Other…’, and, if this is selected, a window pops up allowing you to add a new functional, basis set, etc. which is saved in either the python script directly or in a .avogadro2rc file (if one exists). While it wouldn’t necessarily make things more easy for people who have a lot of extra options (adjusting convergence criteria, specifying multiple basis sets, and playing with print options, for example), it would save the average person the effort of searching through python scripts and editing the code (and potentially breaking it) for what is, essentially, an extra "<method>", line.
It wouldn’t be a great idea to revise the Python script itself (self-modifying code is a big no-no), adding some “Other…” options for functional / basis set seems useful. The original design was “but you can edit the preview below” but I’ve seen that’s pretty rare, and obviously people would like to add some common pieces.
I’ll have to think a little bit about how that would work – right now the scripts don’t know about settings. But they should get some info from Avogadro about previously chosen options (e.g., it remembers that you like def2-TZVP or whatever).