Hi, I tried to use the LAMMPS input file generator but am having issues with trying to run the program. I was wondering if you guys could tell me what I’m doing wrong.
Here is what my input looks like:
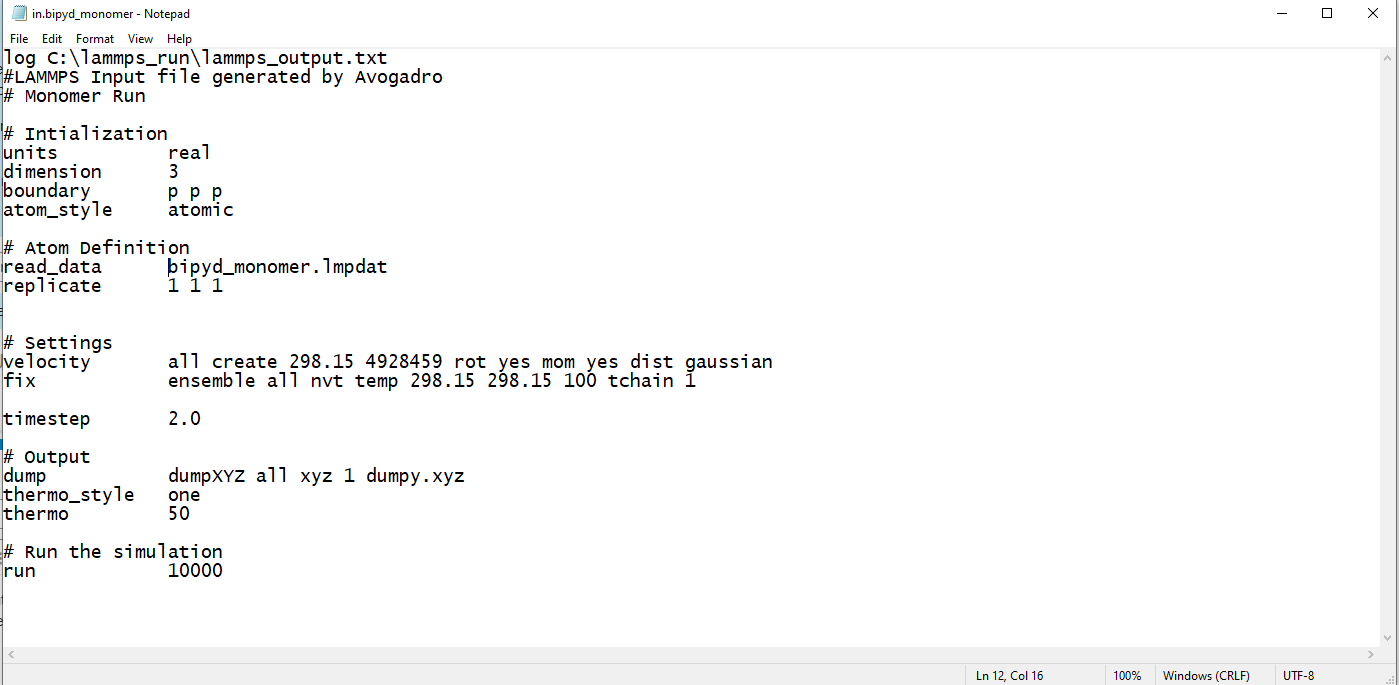
and the error message I am getting says:
ERROR: Cannot open file bipyd_monomer.lmpdat: No such file or directory (src/read_data.cpp:312)
Last command: read_data bipyd_monomer.lmpdat
The data file was created as per the instructions on LAMMPS input for water - Avogadro
I wasn’t sure where to ask for help so I figured I would start with this forum. Thank you for any help!