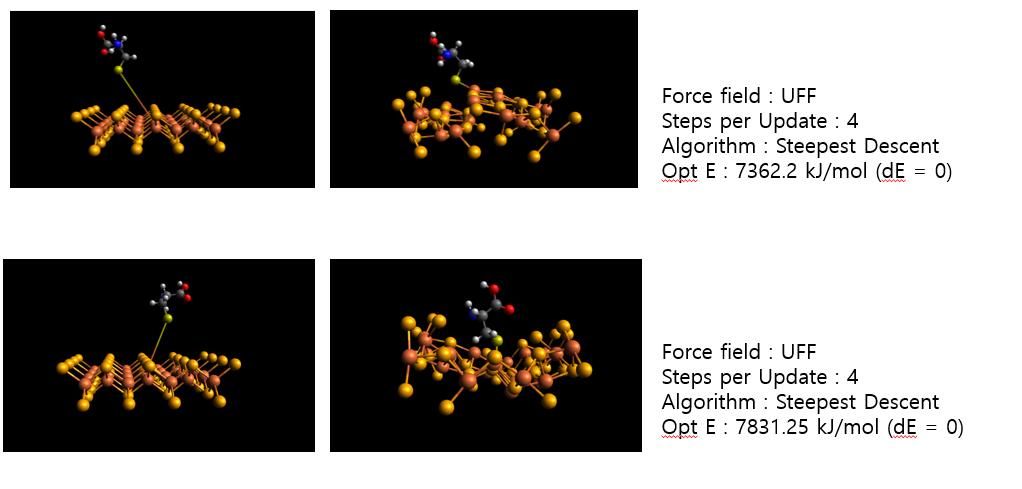
In this case, the bonding and the molecule is same. But only the bond length and molecular direction is different.
In my opinion, I think after the optimization, their energy and the structure must be same as they will both head to the lowest energy level.
But the result is quite different. Do I have misunderstanding?
Could you please tell me the reason why?
Sure, but there are likely many, manylocal minima. Geometry optimization is only going to head “downhill” to the nearest local minima.
You would have to do multiple sampling as far as the position / orientation if you want to find the global minima for your system. (I would also suggest freezing the positions of your surface, because it’s unlikely to warp much.)
@yujin4169 I presume the task ahead of you relates to adsorption / desorption of the organic molecule to the exposed material, possibly vs an oriented surface. That is, because crystalline matter often is anisotropic, there can be significant difference between a (100) surface and a (010) surface in terms of energies to consider e.g. in heterogenic catalysis.
If this assumption is correct, then there (likely) should not be (this much/any significant) conformational change of the Fe-Se bonds of an extended layer/crystalline surface during the approach of the organic molecule. Computational chemists/physicists/material scientists invested in this field of research have a way “to fix and freeze” crystalline surfaces while modelling the approach of a molecular probe in their programs (e.g. Materials Studio). UFF was parametrized to propose reasonable low-energies geometries of small individual, isolated molecules. This is why I would argue that it is not the best tool here.