I believe this to be a bug with Avogadro
Environment Information
Avogadro version: 1.99.0
Operating system and version: Windows 11 Pro Version 10.0.22631
Expected Behavior
Extension>Optimize Geometry should optimize the geometry of the molecule.
Actual Behavior
No geometry optimization occurs, no errors are output, very rarely there is a crash, can not reliably replicate the crash.
Steps to Reproduce
Construct any inorganic or organometallic complex, then attempt to optimize the geometry by either using the menus or by pressing Ctrl+Alt+O.
Current Workaround
In order to get a rough force-field optimization of the complexes I study, I use Extension>OpenBabel>OptimizeGeometry, however the convenience of being able to run multiple optimizations at once in short order with a keyboard shortcut is very nice and so this workaround is just a temporary patch.
First off, I know there were issues with the 1.99 release on Windows. Can you try a nightly build from https://two.avogadro.cc - I’d like to fix a few more bugs and release a 1.99.1 bug fix.
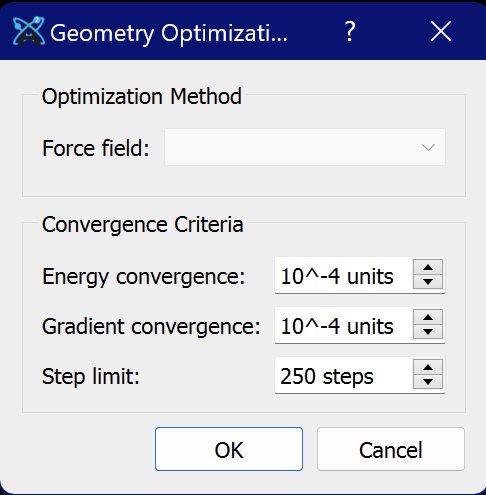
Secondly, if you go to Extensions ⇒ Calculate ⇒Configure… what does it show as options and the currently configured option?
I downloaded and installed the nightly build just now, still no optimization.
For the second part, I have attached a screenshot of the menu that opens in the configure menu.
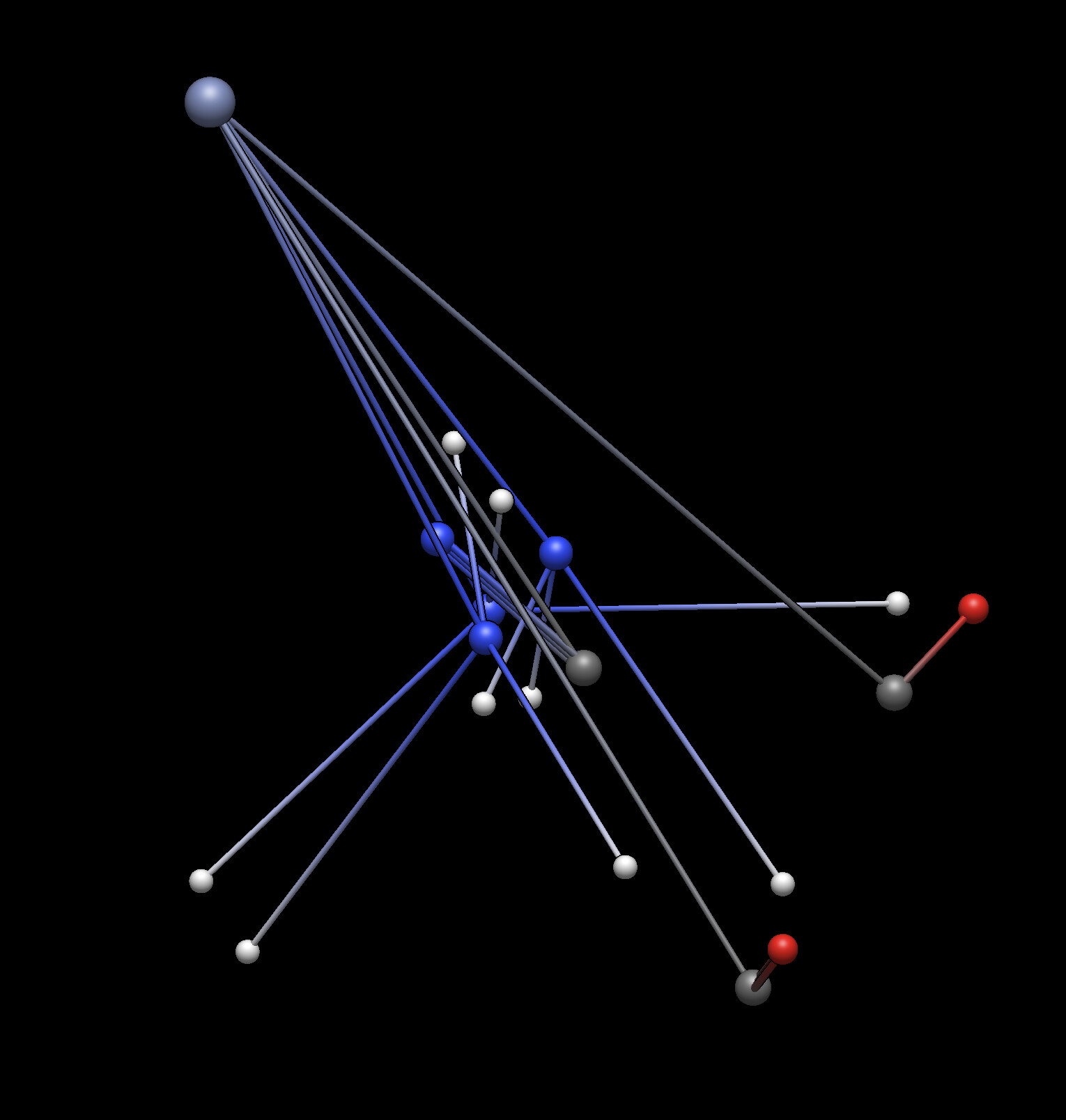
EDIT: After posting this I just absentmindedly tried again to optimize, and it scrambled the geometry. This reminds me of a few months ago when I had a similar issue with the built-in geometry optimization, I think the cause that you [Geoff] suggested was that it was using a basic Lennard-Jones potential and it couldn’t quite handle complex molecules.
On a side note, the optimized spaghetti structures always make me laugh so I’ll include it here.
What is is the import format you use? Can you share the file as an attachment to your question?
I copy-pasted the SMILES strings of of one of the Grubbs catalysts provided by the English edition of Wikipedia, i.e.
Cl[Ru-2](Cl)([c+]0n(-c1c(C)cc(C)cc1C)CCn0-c1c(C)cc(C)cc1C)([P+](C1CCCCC1)(C1CCCCC1)C1CCCCC1)=Cc1ccccc1
into a file Grubbs_catalyst.smi. The geometry Avogadro* provided right on reading this (i.e. no call for an [additional] structure optimization) might not be “the one” of the lowest in energy, but on first sight not too much “off”:
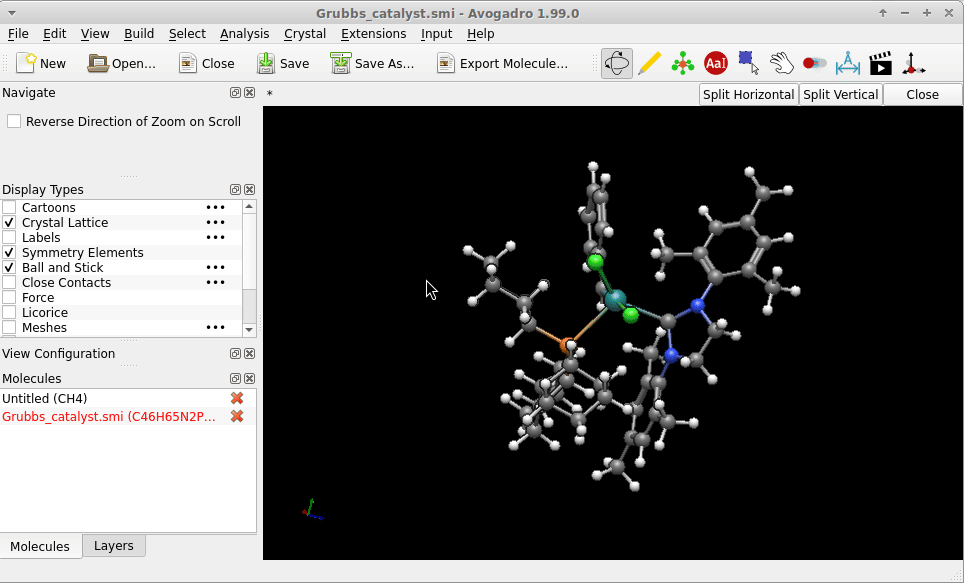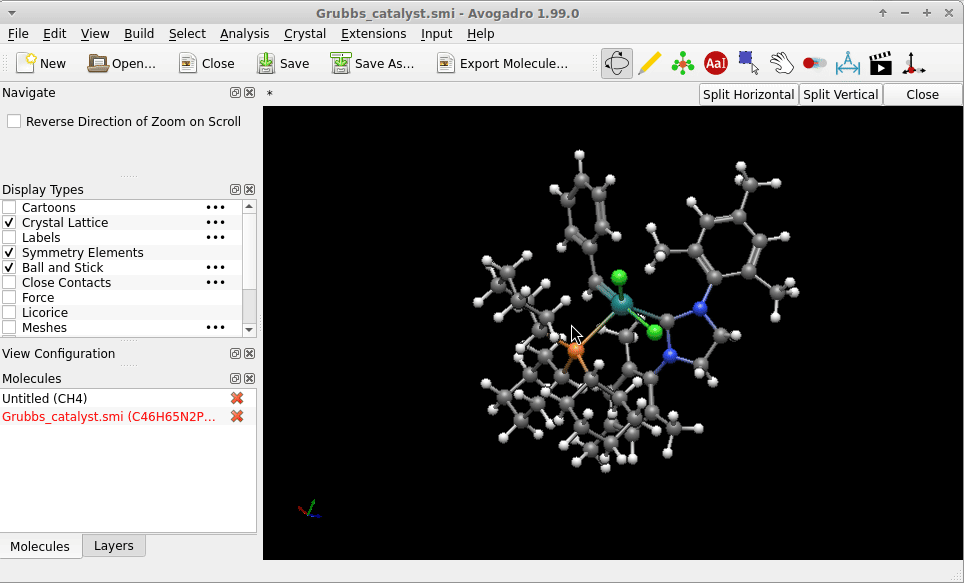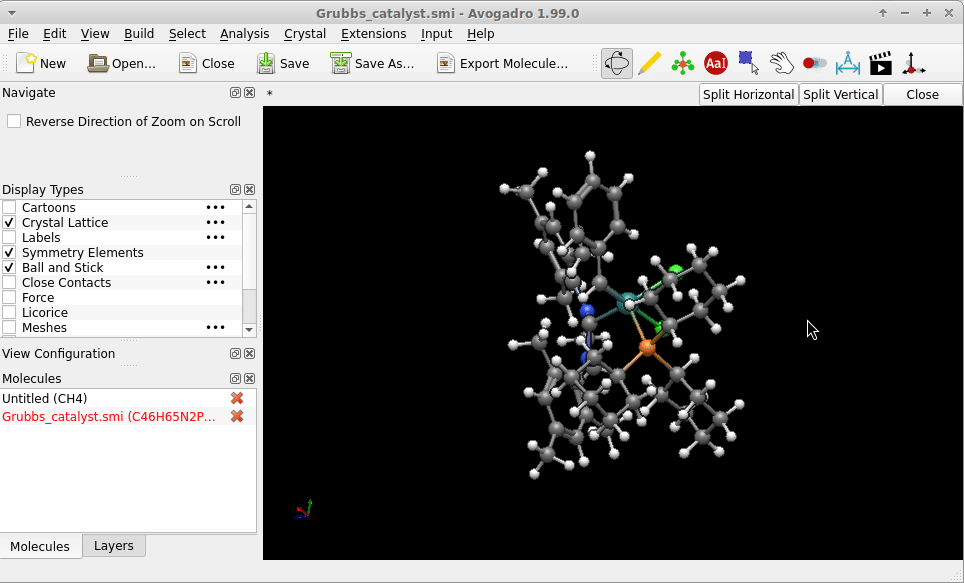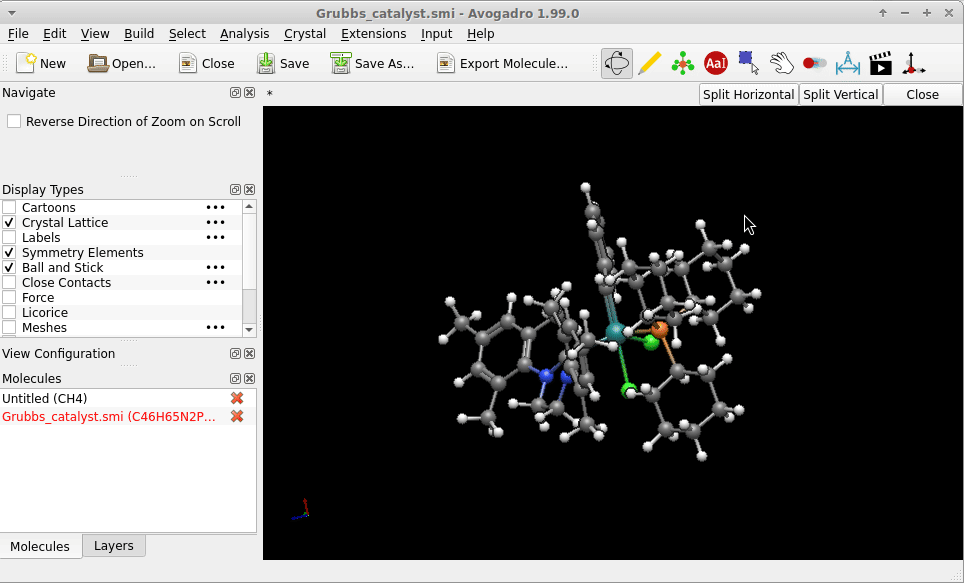
Possibly Wikipedia can provide you additional SMILES strings (there is the chemical structure explorer), or an entry on PubChem (example of a search for Glucose) as a larger database to bypass the problem in case you don’t have a .sdf with atomic coordinates at hand.
* A nightly built Avogadro AppImage for Linux, fetched by 2024-03-11.
I think you misunderstood my issue. The problem I am having is that after drawing a structure using the built-in functions of Avogadro, and then attempting to use the Optimize Geometry function, that last step doesn’t function. It isn’t a matter of inputting a geometry, but rather taking a custom geometry and performing a force-field optimization.
Without prior file of a program (ChemDraw, Marvin; a database), building the molecule in Avogadro, I proceed in lines of the gif attached below:
Can you replicate this workflow* on the computer you use?
In case you already know a bit about the coordination geometry while constructing the molecule model, or want to provide the geometry optimization a hint with a reasonable starting point around the metal; next to the pen is the template tool to accelerate the work (including a couple of ligands to choose from).
I am well aware of how to use all of the features in Avogadro, however the issue that I had was not that I couldn’t optimize the geometry but rather that the feature where you can press Ctrl+Alt+O and have it optimize automatically was not functioning properly for transition metal complexes. This is not a big problem, but I know that Geoff would want to fix it, so I posted about it.
Yeah, I appreciate it.
I can confirm that when I build ferrocene, for some reason UFF (or GFN-FF) is not available. Not sure why.
One of my projects for the next few weeks is to build up a “native” UFF implementation in Avogadro, rather than having to use Open Babel.
I notice File → Import → Download by Name followed by entering ferrocene doesn’t yield Fe packed between the cyclopentadienyl ligands …
That’s not so much an Avogadro issue as a general problem with coordinate generation of organometallic species and the name resolver.
There are multiple “forms” of ferrocene out there (eg in PubChem) which is one reason the new template tool is so useful.
I was thinking about the implementation of FF optimization techniques in Avogadro 1, and I am wondering if you had a reason for changing the continuous optimization that you had used in A1 to a stepwise optimization.
In case my description isn’t entirely clear, consider that in A1 the optimization tool would continually optimize structures until the ∆E is zero. In A2, the optimization tool requires that you specify a maximum number of steps possible until the optimization turns off, even if the optimization hasn’t reached a minimum energy. This is the change that I am curious about.
Yeah, it’s coming. From a coding perspective, the “auto-optimize” tool is harder to debug. A bunch of people were indicating they wanted a way to use other programs / methods to optimize inside Avogadro. You can do that now, and I’m working on ways to improve the speed.
My goal for the summer (and obviously for the 2.0 release) is to get the auto-optimize tool back working. I’ll certainly post when I have some code to test.
For now, you can set up a large number of steps (e.g., 5000) and it’ll keep going until it converges.
Thanks a ton for all the work you do Geoff, I am super grateful that there is someone who is really passionate about this project and willing to put in the long hours!