Good afternoon.
I am unable to open .out files running on gaussian 09 and gaussian 16.
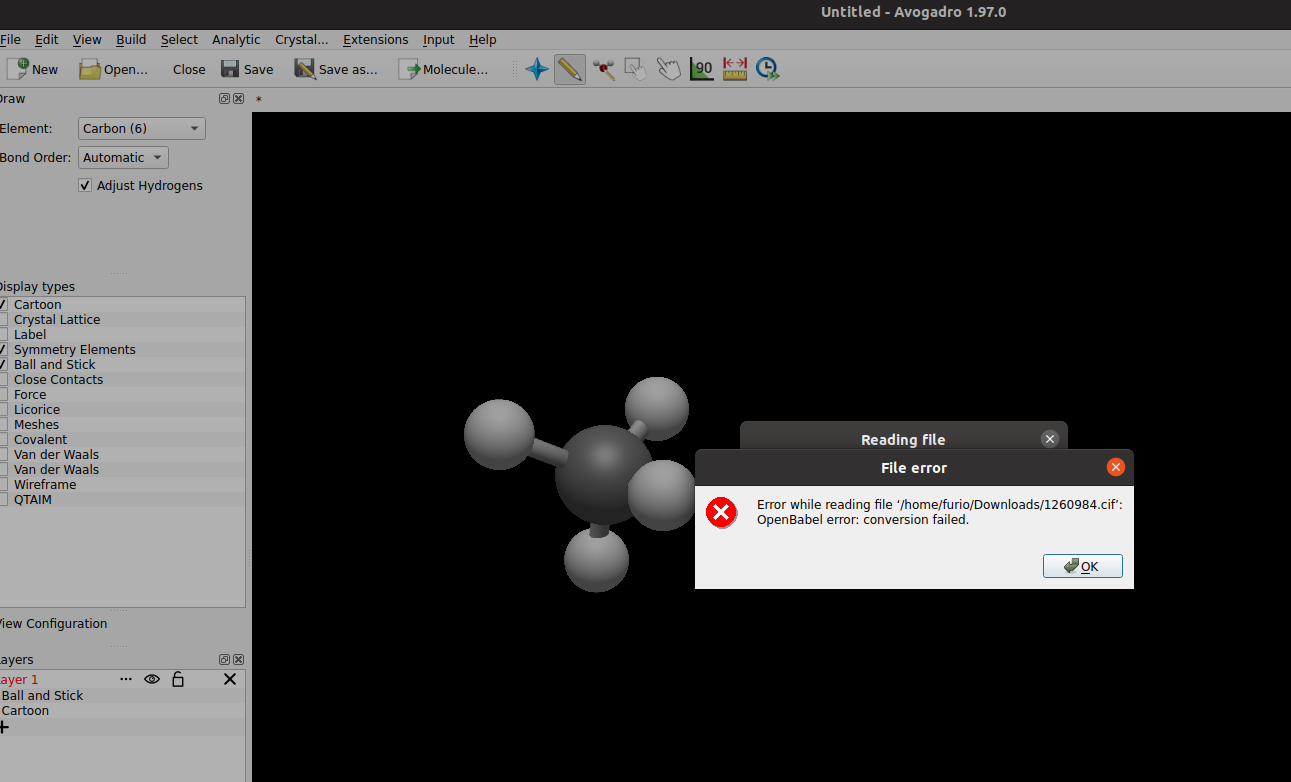
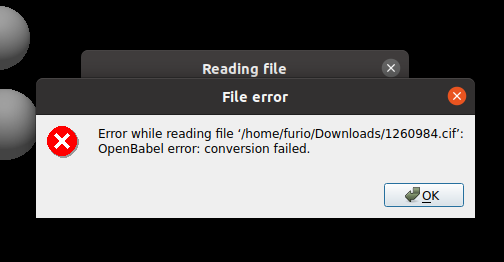
When I open the file I get the following error: OpenBabel error: conversion failed.
Does anyone know how to solve this problem?
Good afternoon.
I am unable to open .out files running on gaussian 09 and gaussian 16.
When I open the file I get the following error: OpenBabel error: conversion failed.
Does anyone know how to solve this problem?
Can you give us a bit more information? What version are you using? What OS? How did you install it? Can you also post a file?
Thanks,
-Geoff
Good morning,
My avogadro updated to version avogadro2, version 1.93.0 and since then I’m having problems that I didn’t have with the old version.
I’m using ubuntu 20.04 OS, but when I used linux mint 20 (Ulyana) I had the same problem.
The problem consists of three main points:
1- The option Extensions >> open babel is disabled.
2- The Quantum option is also disabled.
3- This version of avogadro2 is not opening the .out file generated by gaussian 09 and gaussian 16.
How could I solve this problem?
I installed avogadro2 in two ways:
sudo apt update
sudo apt install avogadro
sudo apt-get update -y
sudo apt-get install -y avogadro
All two forms of installation have the same problem mentioned in the previous msg.
You’ll want 1.94 / 1.95 - we’ve fixed lots of bugs in these versions.
Can anyone suggest please?
I have downloaded from Release Avogadro 1.97.0 · OpenChemistry/avogadrolibs · GitHub the version Avogadro2-x86_64.AppImage on my Linux Ubuntu 20.04.3 LTS. I have then given the executable permission to this Appimage file. After this when I try to open a .cif file I get the following error: OpenBabel error: conversion failed.
Can you please post the file or a link to it? Thanks.
Entry COD 8107379, at time of writing the latest addition to the freely accessible COD is read without a problem (md5sum 8ca3976317b99a4ff61623379c295cb3, Linux Debian 12/bookworm)
which (if you watch closely) includes the assignment of bond borders (phenyl moieties, carbonyl group; display type «Ball and Stick» has a toggle «show multiple bonds»). Prior to an attempt of replication, remove the .txt from the attached .cif below.
8107379.cif.txt (16.7 KB)
I guess the question @Thomas - are you using the Debian (debichem) package? Because @debianubuntupkg is mentioning issues with the AppImage.
My question for @debianubuntupkg … you’re having problems with CIF on that build. Can you read other files through Open Babel, e.g. a PDB file or XYZ file from the AppImage? (In other words, is it a problem with Open Babel import generally with the AppImage or with CIF in particular.)
I ask, because as I work towards 1.98, I’d like to fix the problem.
@ghutchis About half a year ago, I removed Avogadro as offered by DebiChem. Since then, the AppImage of Avogadro is used. This is why I assumed provision of the checksum could be useful to identify the AppImage in question. synaptic’s report is empty
There however is openbabel, installed as provided by Debian’s repositories:
@ghutchis and @Thomas : Thanks for your inputs. They were really helpful.
I am not sure why the .cif file I tried to open did not work. But, now when I opened the .cif files downloaded from COD (Crystallography Open Database) it worked and even the pdb files were also successfully imported.
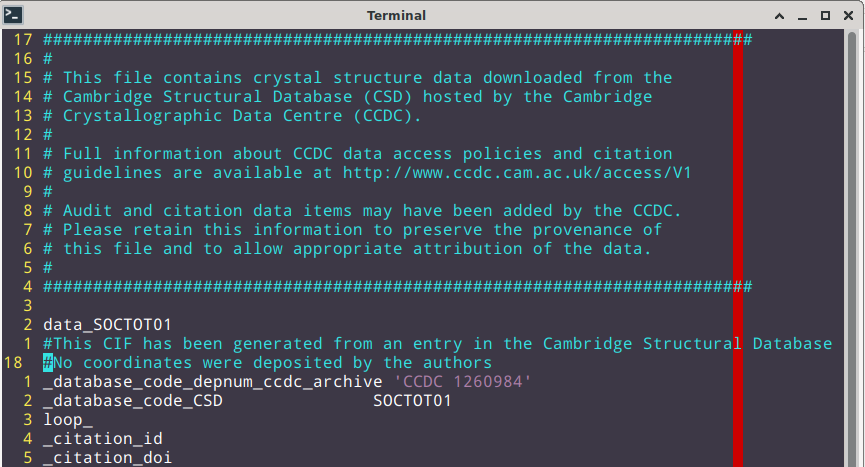
@ghutchis : The earlier .cif file which I tried to open was 1260984.cif which I downloaded from : https://www.ccdc.cam.ac.uk/structures/?
I am not sure why you asked about import through OpenBabel because the error I was receiving was simply when I tried to open the 1260984.cif file via file-open sequential steps and I did not try importing via openbabel. So, I can say the Linux AppImage Avogadro-1.97 version is working fine.
Avogadro 1.97 will use Open Babel to open many file formats, including CIF. I’ll take a look at the CCDC file, since it’s probably an Open Babel error.
The cif file is plain ASCII; any (text) editor at your disposition should be able to process it. Line 18 states the authors did not / did not yet want to deposit the atomic coordinates:
There may be reasons for this. E.g., you want to deposit an entry to the database with an embargo to file a patent application (i.e. entry is present, but not searchable by regular subscribers of the database). However CCDC typically allows this for two years maximum, then you either either retract the entry, state the scientific journal where the structure model is published, or publish the entry as a communication to CCDC.
However here, it possibly is because the structure was a twinned structure. Without going too much into detail (an entry into this large topic), the characterization of the sample can become this complex that to establish a model with a unit cell / lattice vectors a, b, c and space group symmetry of the unit cell without determination of atomic coordinates already is an achievement. (This can happen regardless of equipment and computer programs to process the data at hand.)
Contrasting to the CCDC, it is explicitly permitted to download individual entries, or the whole COD altogether to complement a (subscription based) query in the CSD file. (No database contains all data, though there are overlaps.) To query, you can use their search mask, the one by crystallography.io, or use it as a MySQL database. At present, the COD holds 7 entries about C60 in six different space group symmetries. Entry COD 9011580 possibly is a match (though the model is based on data recorded at 5 K) with the symmetry from the CSD file.
And while it’s not at the top of my list, I want a “Search COD” interface in Avogadro 2.0.
I think my take-home from this thread is that:
Jmol possibly has some advance (compared to Avogadro) to interact with crystal structures in part by their «Crystal Community» and e.g. Robert Hanson’s tutorial-like publication in 2010 in J. Appl. Cryst. about visualizations. (Still, CCDC’s Mercury often is a prompt association when “reading a .cif from the CSD file” is the next task ahead; not only because of the integration with the subscription based database.)
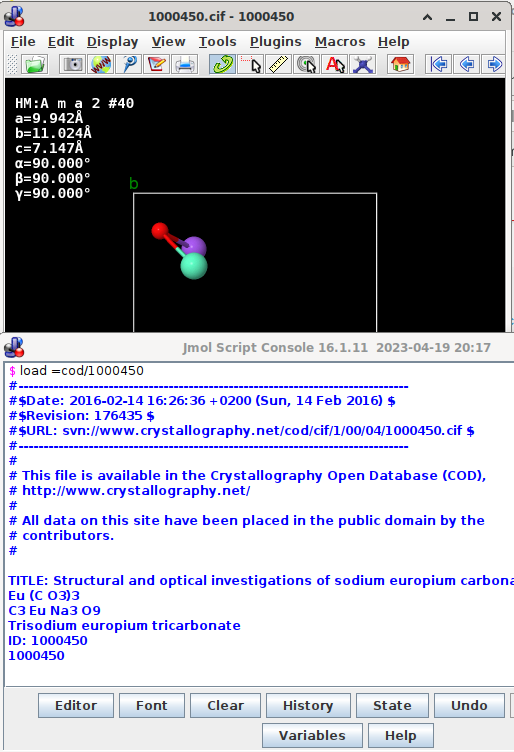
In Jmol, a call to the COD, a call from the program’s terminal (File → Console) in line of
load =cod/1000450
issues a request predictable in pattern to
svn://www.crystallography.net/cod/cif/1/00/04/1000450.cif
as an external database (=) which happens to be the COD (cod, an overview of sources accessible to Jmol) to pass three layers of folders / levels of hierarchy ahead of accessing the .cif in question. Maybe this information can help to set up an interface between Avogadro and the COD.