The goal is to add hydrogens to the ligand structure.
What am I doing:
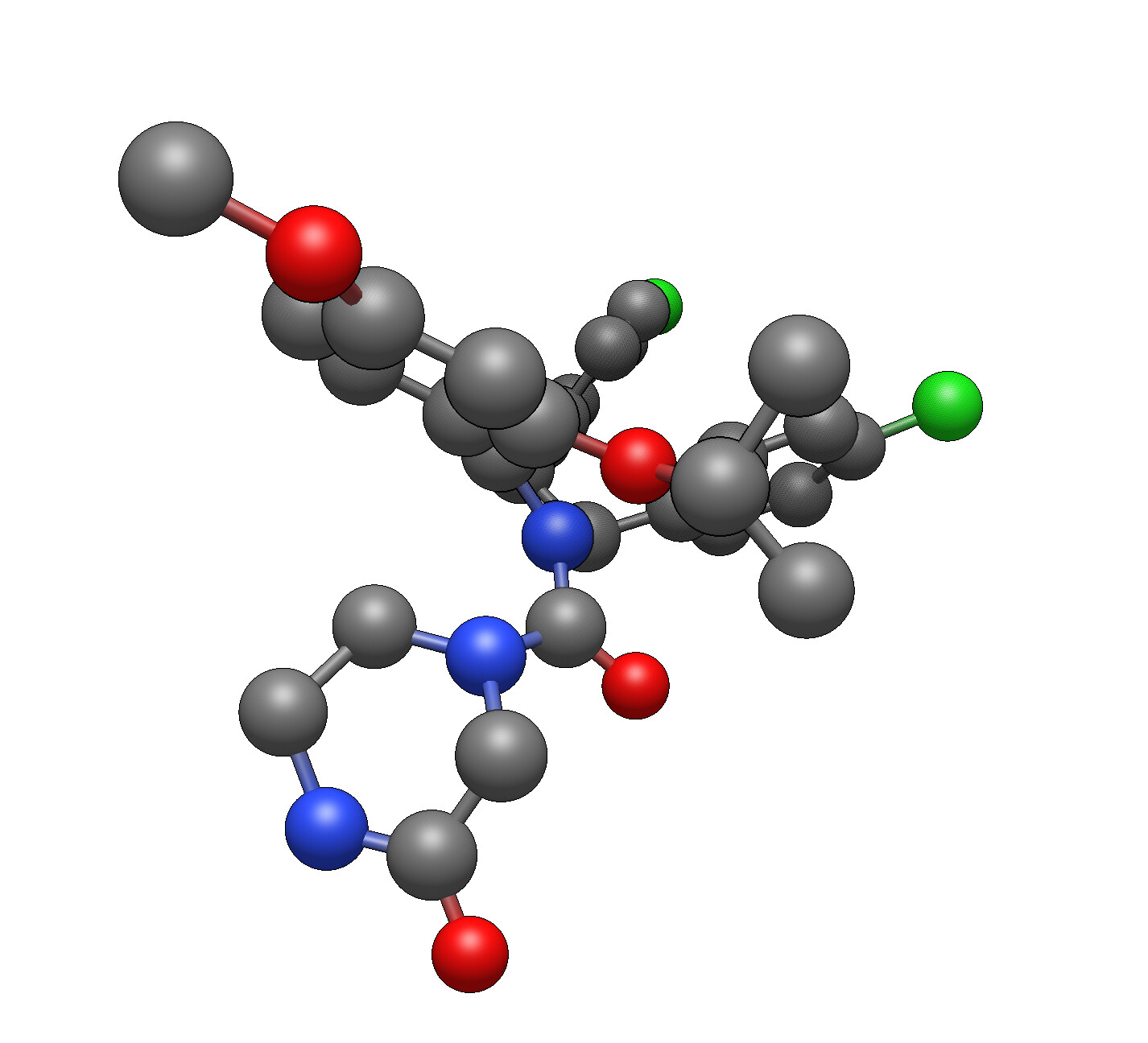
– Downloaded the structure from the PDB database.
– Cut out ligand molecules.
– Saved it in a new text file ligand.pdb.
– Open ligand.pdb in Avogadro 4J3E_ligand.pdb (3.2 KB)
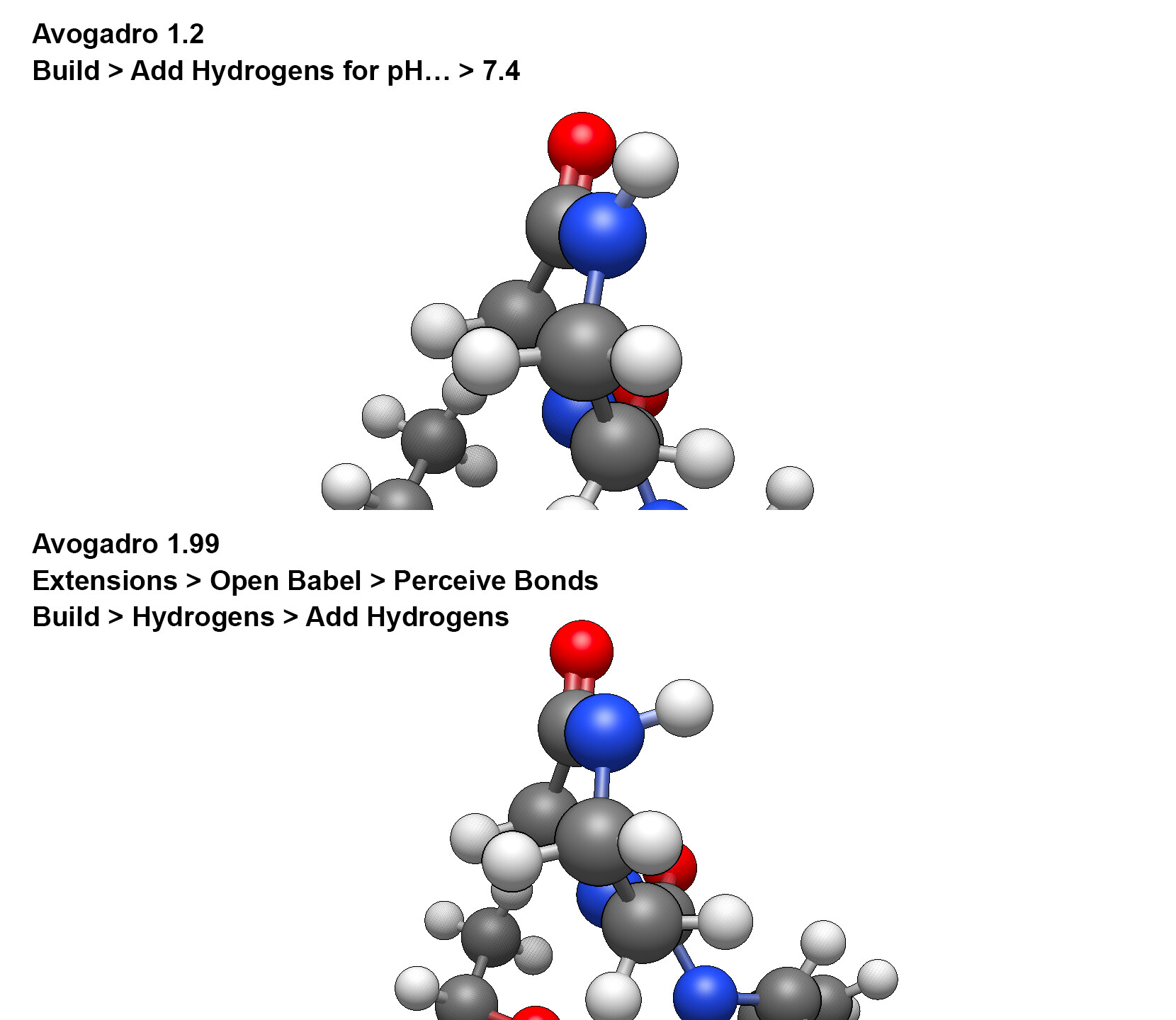
The first version of Avogadro (Version 1.2.0) did the following steps to add hydrogens and arrange bonds
– Build > Add Hydrogens for pH… > 7.4 > OK (bonds were created and 33 hydrogens were added in the MMFF force field)
The second version of Avogadro (Version 1.99.0) attempts such
1st try
– Build > Hydrogens > Add Hydrogens > ERROR (double bonds are not established and 54 hydrogens are added, to all possible atoms)
2nd try
– Extensions > Open Babel > Add Hydrogens for pH… > ERROR (3 hydrogens are added, while if you run obabel from the command line, 33 hydrogens will be added)
3rd try
– Extensions > Open Babel > Perceive Bonds > OK (bonds were created)
– Extensions > Open Babel > Add Hydrogens for pH… > ERROR (nothing is added at all)
4th attempt
– Extensions > Open Babel > Perceive Bonds > OK (bonds were created)
– Build > Hydrogens > Add Hydrogens > OK (30 hydrogens added)
Question 1, why can’t I add hydrogens using Open Babel?
Question 2, in the 4th attempt, what force field is used when adding hydrogens?
P.S. The difference between 30 and 33 adding hydrogens is the correct identification of double bonds.
– When using the first version of Avogadro, you need to establish one double bond with your hands.
– When using the second version of Avogadro, the double bond is automatically established correctly.
Have a nice day, your answer also ended up in spam. I just found it now
There’s no “force field” for adding hydrogens. There are a set of empirical rules.
Okay, got it
It sounds like there’s some issue with the bond perception. Do you have an example of what the bond orders should be?
I’m trying to open a ligand file cut from the PDB complex (i.e. it doesn’t have a connectivity table).
For the first version of Avogadro (version 1.2.0) this was not a problem, you could create bonds and add hydrogens with one button (Build > Add hydrogens for pH… > 7.4 > OK).
For the second version of Avogadro (version 1.99.0), it’s impossible to add hydrogens using OpenBabel (I tried it in different ways, see the previous letter).
It sounds like 1.99 doesn’t get the correct bond perception, which is why you’re having issues with adding hydrogens. While I can guess some of the rings, it would help considerably to know what exactly the ligand (and bond orders) are.
After the restoration of double bonds using OpenBabel (Avogadro 1.99), this ring is determined correctly: C=O and nitrogen with a single bond.
I am attaching the ligand files after restoring the connectivity table and adding hydrogens below.
We found out about the definition of double bonds. If a connectivity table is not specified in a molecular file, then Avogadro 1.99 does not understand it.
Next, I use Avogadro’s 1.99 functions to restore it. They work correctly:
– Extensions > Open Babel > Perceive Bonds > OK
However, further addition of hydrogen atoms (after the double bonds have been restored) using OpenBabel (Avogadro 1.99) does not work:
– Extensions > Open Babel > Add Hydrogens for pH… > ERROR (nothing is added at all)
Using Avogadro’s 1.99 internal module to add hydrogens (after the double bonds have been restored) works relatively well:
– Build > Hydrogens > Add Hydrogens > OK (30 hydrogens added).
Let me try again to explain my question about the force field used when adding hydrogens. Earlier you said that the force field has no effect on the adding of hydrogens.
However, if we look at the results of adding hydrogens using two working methods, we will get a different arrangement of hydrogen relative to the six-membered heterocycle.
The correct placement of hydrogens is carried out in the top picture (Avogadro 1.2). I checked this using optimization in various 3D editors.
To make Avogadro 1.99 work as well as the old one, it would be good to make one of the fixes
– fix OpenBabel module for adding hydrogens (it doesn’t work now)
– fix the operation of the internal hydrogen addition module (currently hydrogen is added to the wrong place)
If I’m correct, the only issue is with that nitrogen atom … it should be sp2-like because it’s next to a C=O, right? That’s an easy bug fix. That’s not a “force field” although I understand your comment, because it’s basically an atom type perception.
Or is there another mistake in the hydrogen addition?
I don’t know, I’ll take a look. It could be an easy fix … or it could require some work in both Avogadro and Open Babel.
The change in hydrogen geometry at that nitrogen is ready … I just want to make sure there isn’t something else.
Yes that’s right. Nitrogen in sp2-like because it is bound to C=O. Therefore, the geometry is flat and hydrogen is placed in a plane with carbon and oxygen. I don’t see any other protonation errors here.
Thanks for the instant fix! I’ll definitely check it one of these days.
Thank you, it’s not urgent.
I have already checked all the settings of Avogadro 1.99 for connecting the OpenBabel module, everything is installed correctly and some of the OpenBabel functions work well (Configure Force Field, Optimize Geometry, Perceive Bonds, Remove Hydrogens), but so far there is no way to adding hydrogens.