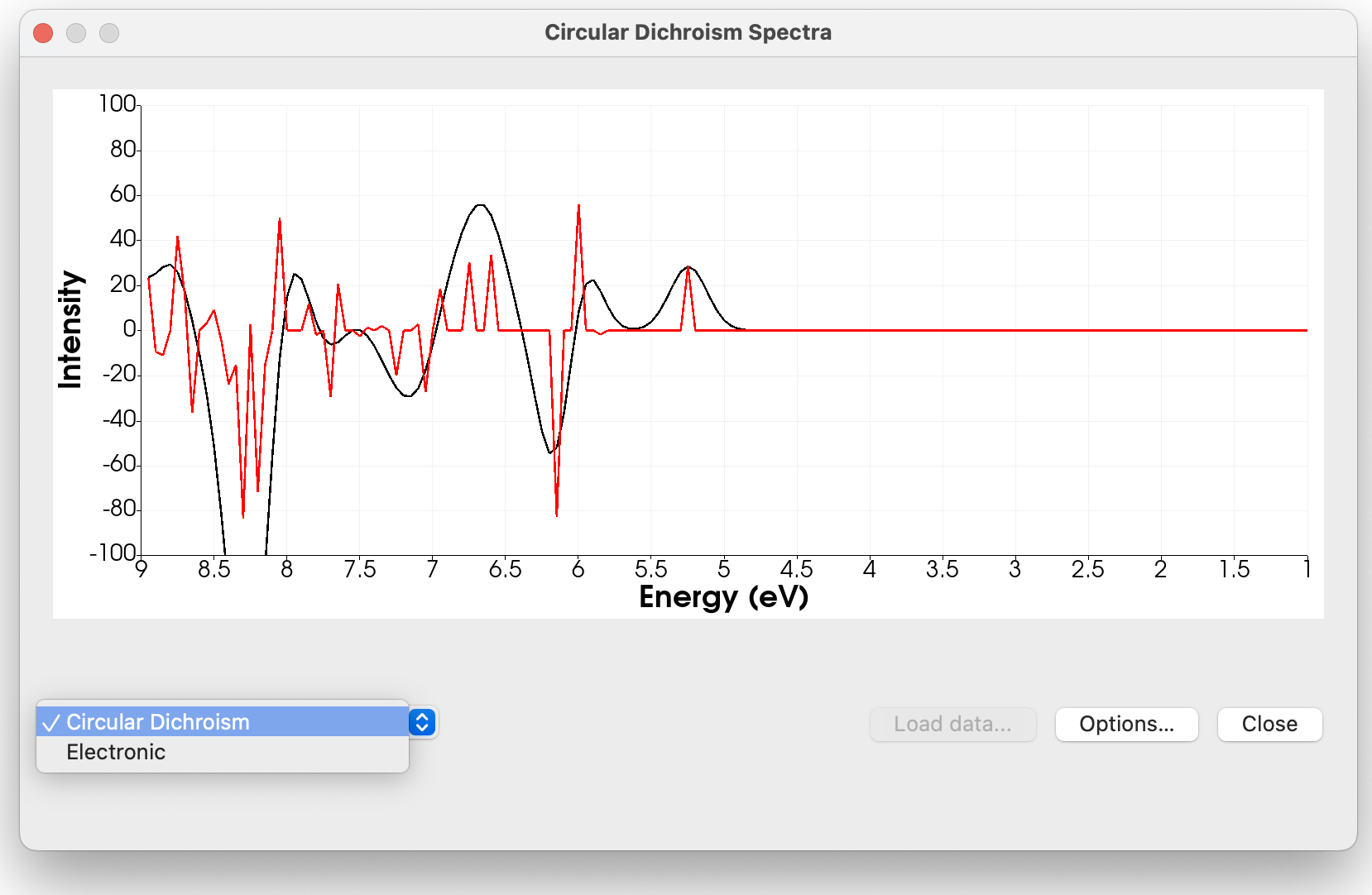
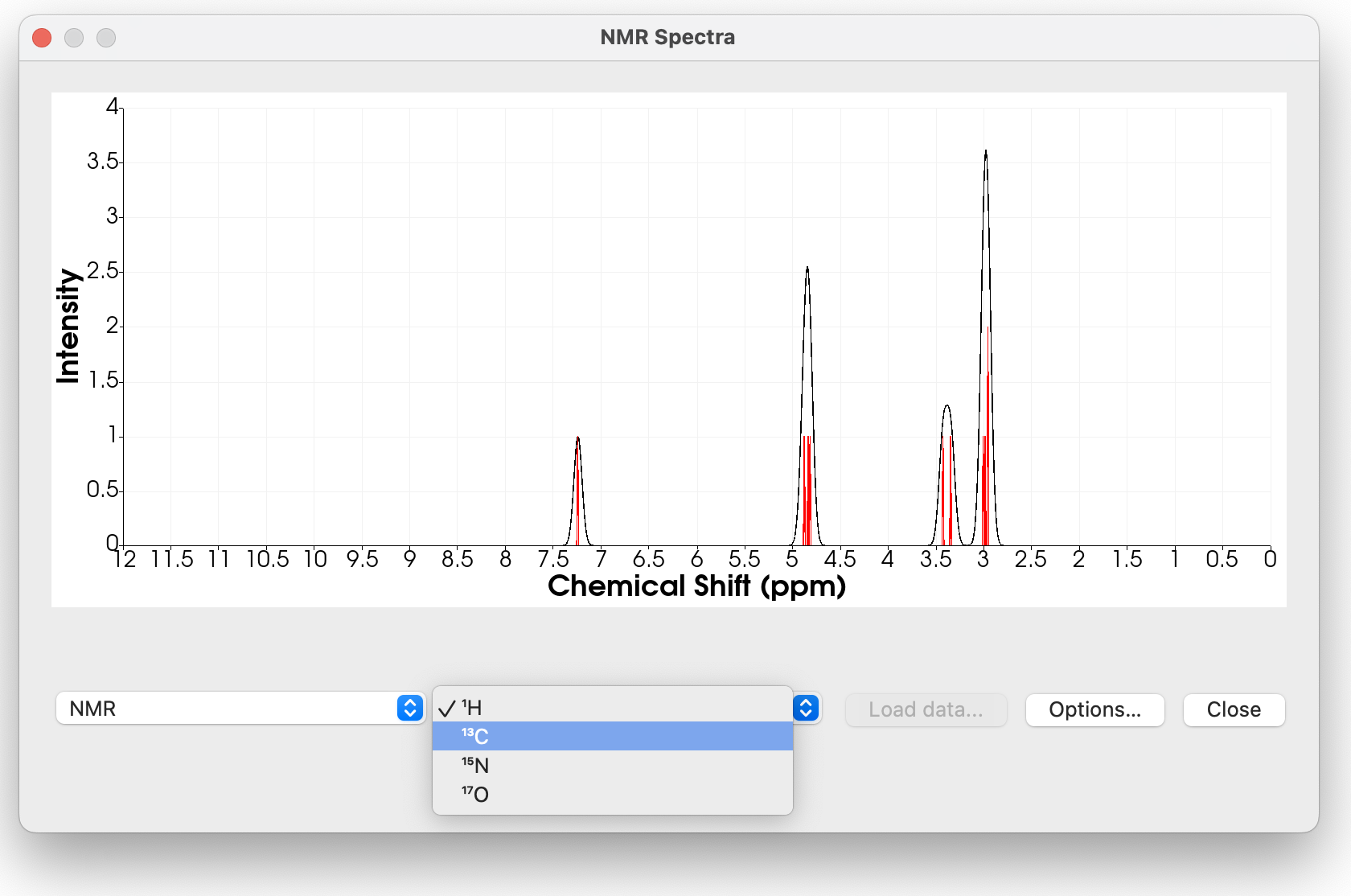
The NMR spectra has some approximate default shifts for 1H, 13C and a few other nuclei (Si, F and N) – at least where I could find the standard. Of course it’s important to calculate the shift for TMS or similar (\ce{CFCl3} and \ce{CH3NO2}) using a particular method, but I felt it was important to give reasonable defaults.
Anyone who has ideas on improving the NMR spectra, please let me know (e.g., handling spin-spin coupling calculations, etc.)
@ghutchis I speculate the enthusiasts around Luc Patiny (EPFL Lausanne/Switzerland) could share ideas/hint to implementations based on experiences with and around nmrium.org (its GitHub repository). Are some of them subscribers to this discussion board?
My understanding (having played with NMRium a bit for teaching) is that it’s primarily for processing experimental spectra. And yes, some of that is on the long-term TODO (e.g., to help match up experimental and computed spectra). A student was also asking me about computing 2D spectra.
As I mentioned, one question I have is for less-common NMR elements. For example, the reference for ^{31}P is “85% \ce{H3PO4} in aqueous solution.” Experimentally, that’s fine. It’s a bit harder to know how to run a DFT calculation - should I use the protonated \ce{H3PO4} in an aqueous solvent model?
I’ll be reaching out to a variety of folks once the release is finished. I’m sure we’ll get plenty of feedback.
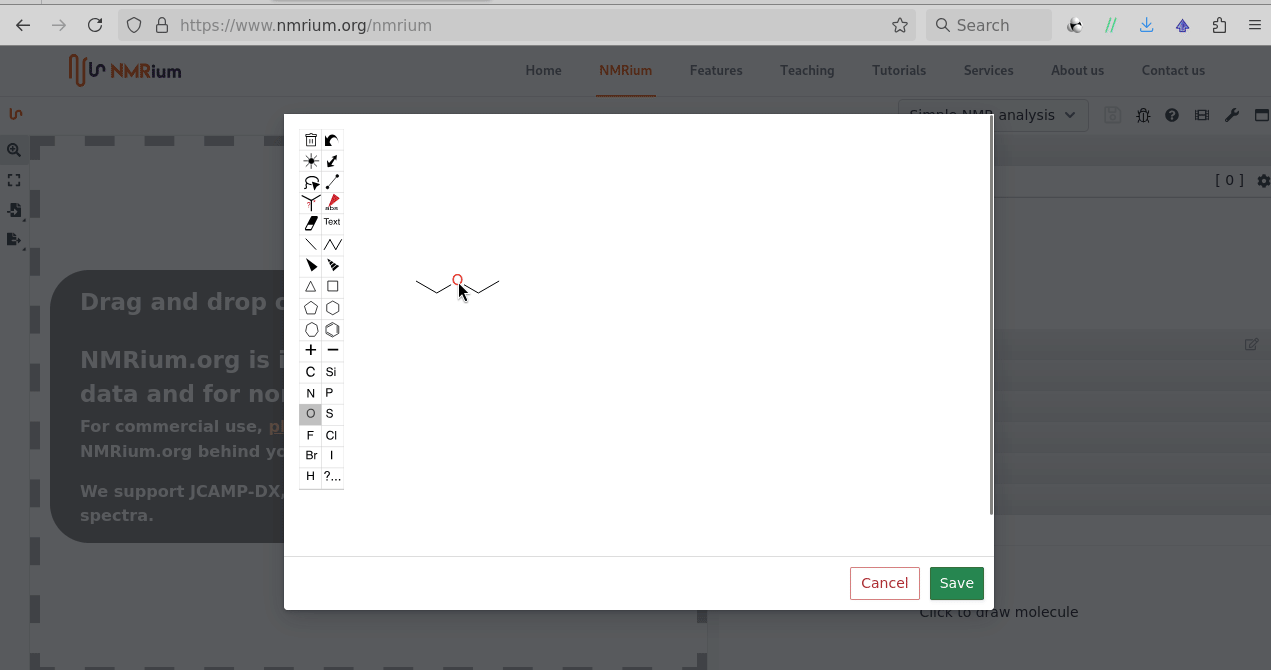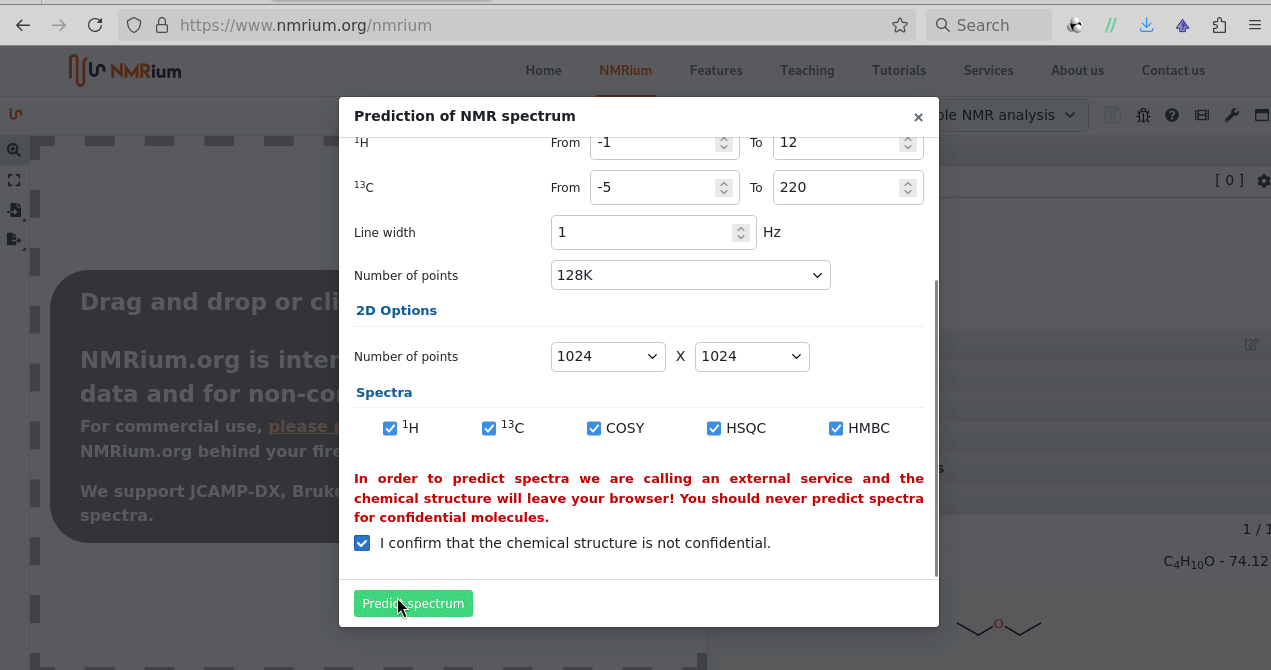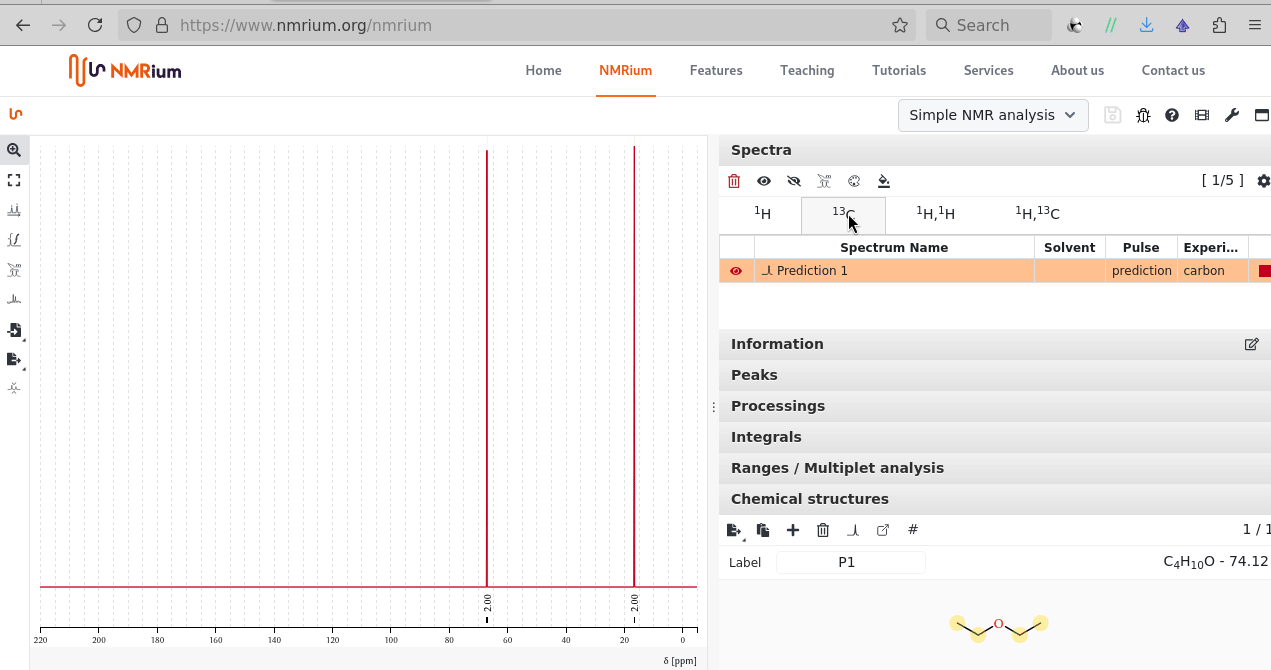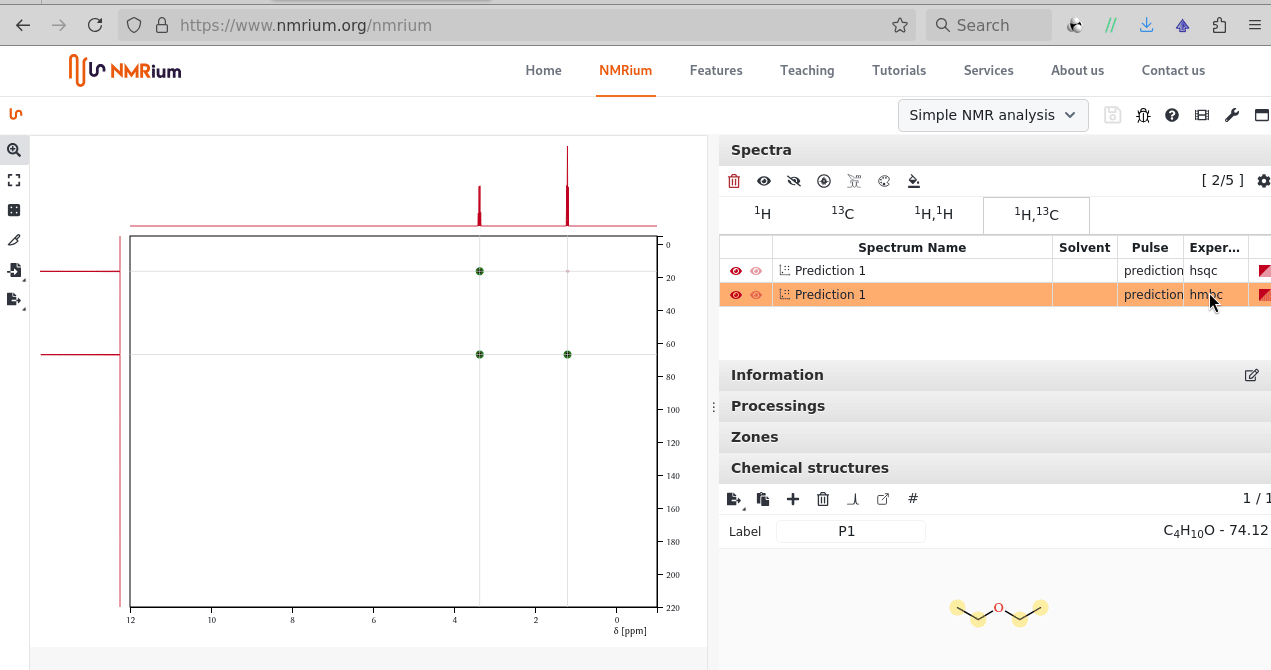
There is sketcher; after the molecule is shared an additional peak icon allows to proceed with a prediction (1H, 13C; 1H-1H COSY, 1H-13C HSQC, 1H-13C HMBC) one can equally process by zoom, drag, integration:
It is better than ChemDraw, ChemDoodle; nmrshift.db? Well, it surely fits a gap, the question then is “better than … in terms of which scale?” (Likely yes for lowering the barrier of learning if the school doesn’t share a site license for ChemDraw/ChemDoodle.) But the prediction doesn’t know (yet) about the couplings like 31P vs 1H/13C seen for P-organic compounds (the HWE precursors, or PPh3 ligands alike), or couplings because of 19F’s presence.
I think that for NMR chemical shifts from external calculations (e.g. DFT) that there should be a spot to specify a TMS chemical shift value to use as a zero point. Or really just specify a spot for a reference value, and just say like
Reference Value (e.g. TMS, CFCl3, H3PO4):
Also, if anyone is curious, I find that for molecules with relatively low degrees of freedom that using KT2/pcSseg-2 generally works, but with molecules that have large amounts of conformers a vibrationally averaged spectrum usually is required (probably with the same KT2 method).
If you click “options” you can enter the reference. Depending on the program, method, basis, etc. reference values can shift by a lot. But I wanted something in the ballpark, so these are from Orca B3LYP def2-TZVP.
Clearly the user manual needs to mention how to calculate reference peaks, and IMHO include links to the files for people to use as starting points.
I agree that a lot of properties / spectra need conformer averaging (and isotopes), but that’s probably a post-2.0 feature to be honest.