I tried to scroll through all the tables but I do not know for sure if I have seen this one.
ch for a CH group
In (heterogeneous) catalysis, you have a lot of surface intermediates like CH, CH2, CH3, CH4, COH, HCO, HCOH, H2CO, H2COH, H3COH, COOH, HCOO, HCOOH, a bent CO2, NH, NH2, and NH3. Some of these are already mentioned, but not all.
Some of the intermediates are not as common for molecules, but I wanted to mention them when Avogadro intends to appeal to heterogeneous catalysis researchers.
If you’d like to add a bunch of these to the fragments repo you can create a directory like surface_fragments or similar and upload the cjson files. In some cases, it’s easier to generate from SMILES, but I suspect a few of these need formal charges, radicals, etc.
The main requirement is to have the attachment point as a dummy atom (element zero). There are a few ways to do that, e.g. using the atom coordinate editor and changing the atom in question to Xx.
We’d be happy to help. Both @Thomas and @matterhorn103 have contributed to the fragments, and contributions are definitely welcome.
How the current fragments work is by replacing an atom right? This would probably not be suitable for surface species.
Because you want to add your surface species above certain selected atoms with a variable distance. For example if you have extended surfaces like (111) and (001) you would add species above different atoms and with different distances to the surface.
I guess you would need to first add a dummy to than replace by the surface fragment. But maybe I do not fully grasp the fragments menu.
Sort of. As it stands, fragments are added in two possible ways:
-
You click on either a hydrogen atom or a dummy atom, and it is replaced with the currently selected fragment
-
You click on empty space, and the fragment is added at that position (with the centre of mass being positioned where the mouse cursor is)
Ideally for surfaces you would click-and-drag from one of the surface atoms, right?
This is incidentally one of the reasons that I proposed combining the Draw and Template tools – you would then have the flexibility of the Draw Tool when adding a fragment, and click-and-drag would be a natural part of it.
Specifically for CH and CH2 though, these are very simply groups, so why do you need to add them with the Fragment Tool? Can’t you just click-and-drag with the existing Draw Tool to insert them? If you draw a short bond distance, the bond order and hydrogen count will be appropriate.
For “free” molecules which can somewhat relax, one can draw them with an editor, and continue with the exported a slightly optimized .mol as in the section of creating new ligands here. But since molecules’ bond lengths and geometries often differ if in solution vs the ones adsorbed, perhaps a dedicated library of motifs for heterogeneous catalysis (as an optional plugin?) suits the particular needs better.
I agree with @Thomas.
Usually, the heterogeneous catalysis systems are significantly bigger than molecular ones. If you add a CH or CH2 by hand as @matterhorn103 suggests, you have to spend some compute time on optimising this adsorbate.
A fragment with a set bond length would also not be perfect for every system but would at least be close to the final solution. It would also be a consistent way to build your intermediates across different systems
I guess this is kind of getting away from the Template Tool Key Shortcuts? Should I start a seperate topic?
I created a new topic. As I said, the point of the template tool is a molecular equivalent of a “stamp tool” in graphics programs.
If you’ve got a set of useful / interesting fragments, I’d welcome the contribution (e.g., in a new subdirectory of fragments/groups. One thing to consider is that the current tool builds off of replacing hydrogen atoms. On a slab surface, you might want somewhat different geometries (e.g., atop vs. bridge vs. 3fold or 4fold).
So this might be better suited to some sort of command, in which you select the surface atoms, pick the group, maybe the binding distance and the binding geometry?
I guess my question goes towards whether this is a mouse tool or something useful for building good geometries (i.e., a command on the Build or Crystal menu).
A side question is whether it’s useful to bring back the slab creation tool or simply providing scripts to use ASE or Pymatgen to generate surfaces.
@joerivan The library of fragments for surface catalysis might call for an extension how Avogadro2 stores the information in the cjson format.
It is possible to freeze selected atoms, their relative position remains unaffected by an energy optimization. Internally, the cjson file labels them only as selected. Yet it is not evident if these groups remain locally rigid (a bit like rigid groups in PXRD) if one/multiple of these particular fragments are used to build new structures.
Small demonstration: intentionally distorted ethanolamine, frozen along the two bonds of \ce{C-O-H}:
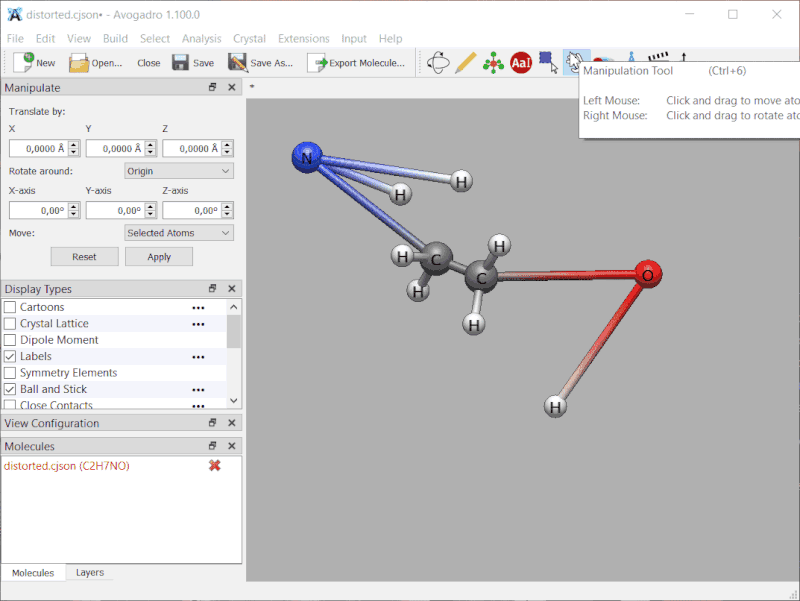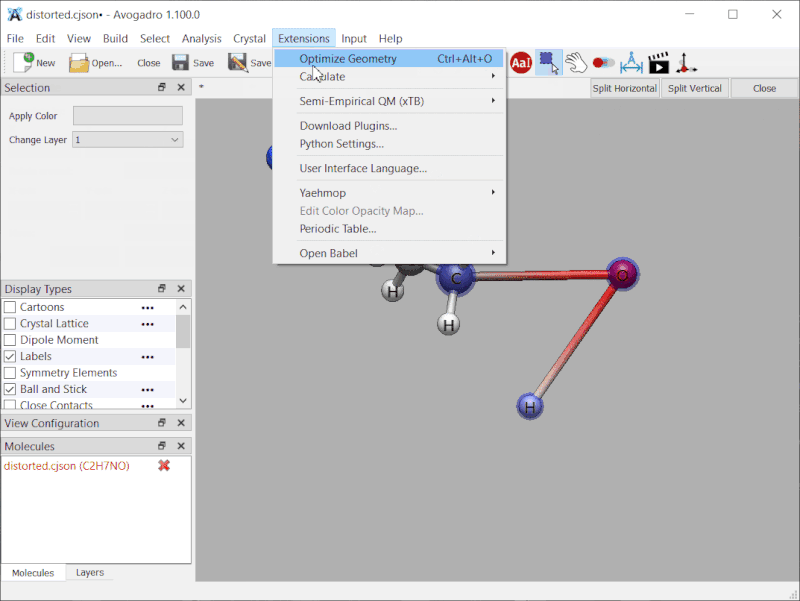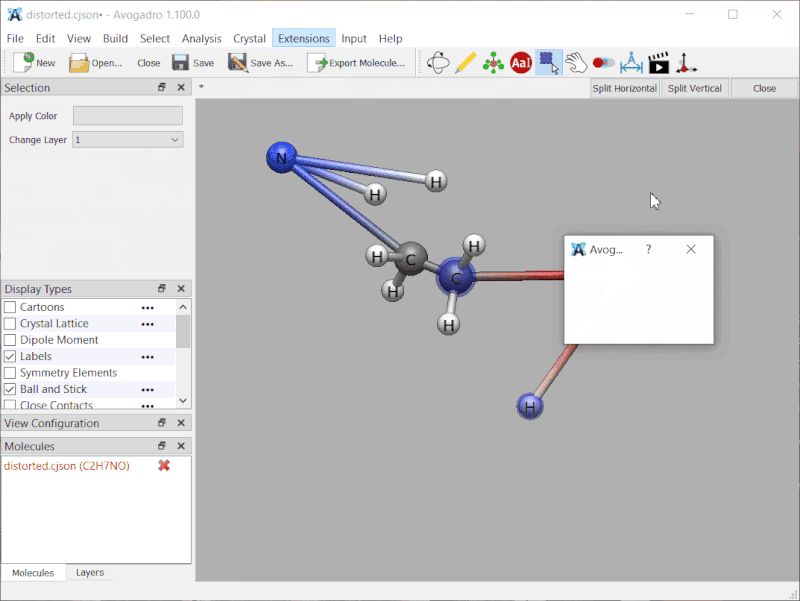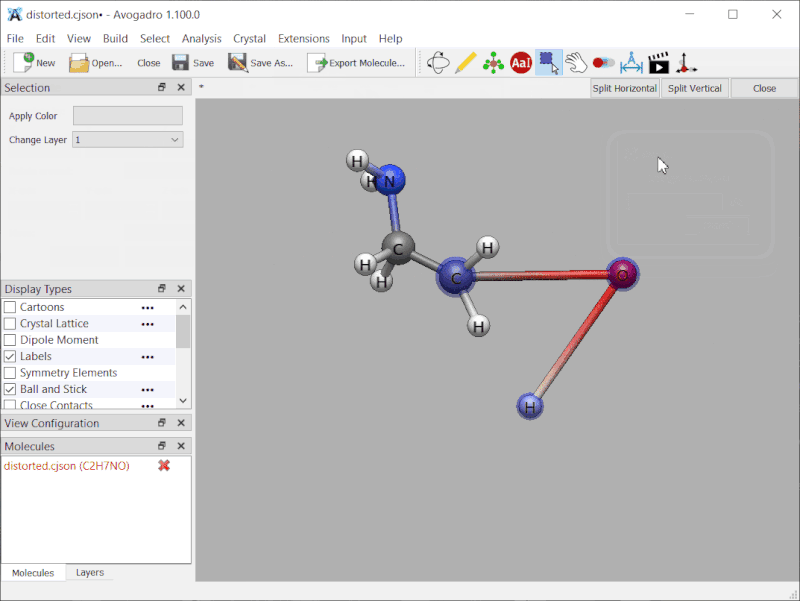
No, it’s just that I forgot to add the frozen coordinates (and soon the constraints) to the CJSON read / write code. I’ll mark that on my todo - probably by the weekend.
Several people have asked about having locally-rigid structures for optimization. My suggestion is a “Fuse Selected Atoms” command that creates a rigid body. (And obviously an “unfuse” version too.)
Fusing atoms is a bit different from freezing them - they can still translate and rotate during the optimization (e.g. via electrostatics / VdW interactions).
Internally, the command simply creates a set of pairwise interatomic distance constraints. For example in water, you’d constrain the H-O bonds but also the H-H distance to retain the angle.
I’ll see if I can add the fuse command tomorrow if there aren’t too many meetings. The hold-up on constraints for geometry optimization has been the UI more than the underlying code.
From my side, the construction of the fragment library will have to wait, but I think I will be able to contribute later this year.
A separate command (something like Add Adsorbate) in the Crystal or Build menu would make a lot of sense. Besides selecting the atop, bridge, 3fold, and 4fold atoms, you would also need to define what direction is normal to the surface.
The command that @ghutchis is sketching would indeed provide all the nobs you would like to turn, I think.
Avogadro would probably not be the main way people construct surfaces. Providing an ASE script is probably best. Another workflow is that people like to optimize the bulk first and then cut the surfaces. ASE can also help with this.
No worries about timing.
I think in many cases I’ve seen, since the slab is more than one layer deep it would be fairly easy to guess the normal. (Graphene and other 2D materials excepted.) We actually already have a command “Add Perpendicular” used for generating ligands like \eta^5 Cp rings.
I think I had an ASE plugin somewhere. I’ll see if I can add a “generate surface” script to it.