Hello I’m trying to display the information from population analysis done on orca.
Particularly Mulliken charge analysis, I’m attaching the output file from orca. Avogadro currently is not reading it, just shows the partical charge in labels.
Is there a way to show there, there is a post on the same topic but for guassian.
Well, you calculated the Hirshfeld charges, which is what it’s showing as atom labels in the latest version of Avogadro2. (You didn’t say what version of Avogadro you’re using.)
At the moment, there isn’t a great way to pick which (of multiple charge models - your file also has Lowdin charges) partial charges to show as atom labels.
So yes, it’s displaying the information from the population analysis, just not from the Mulliken charges. (And out of curiosity, why do you want the Mulliken charges if you calculated the Hirshfeld charges?)
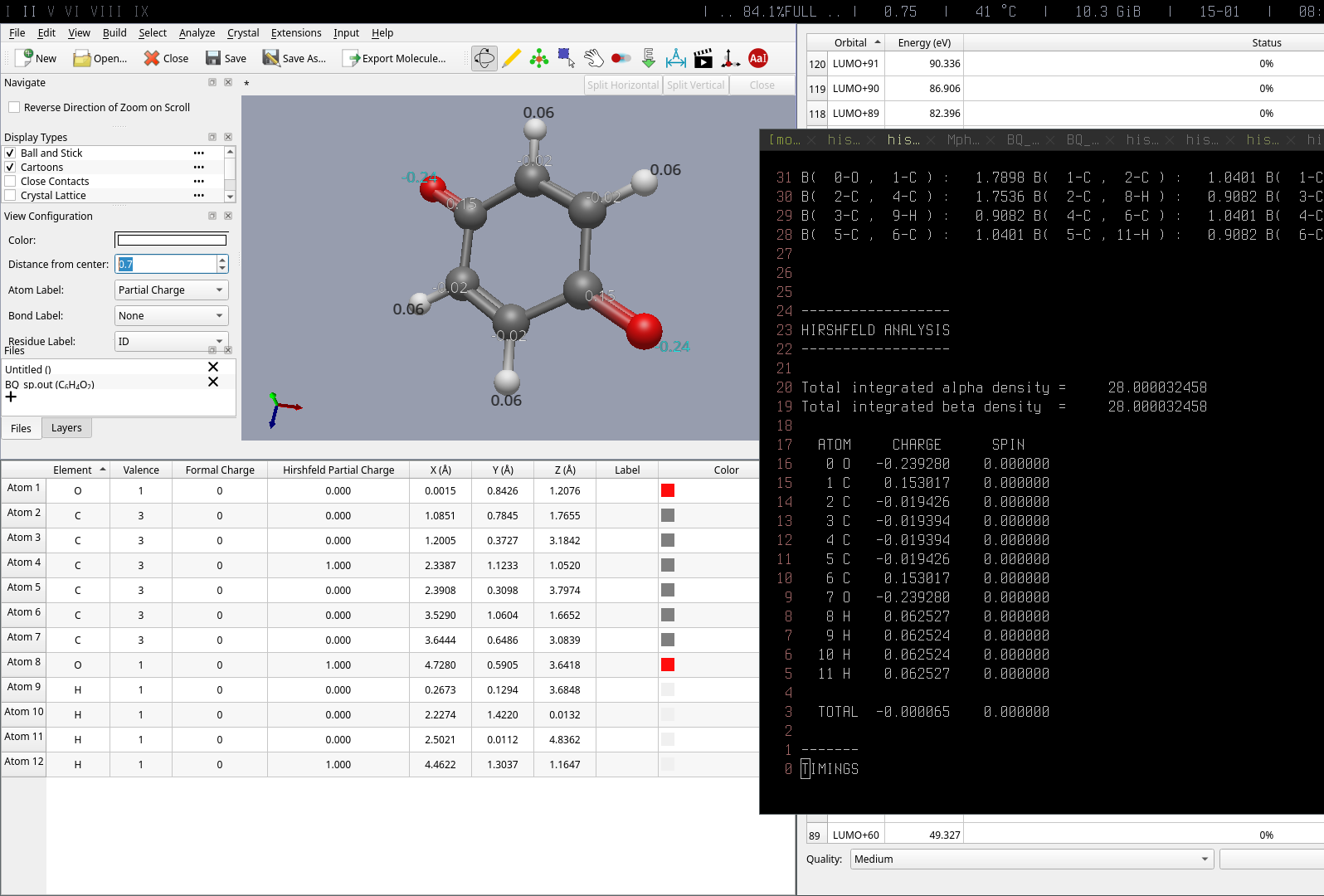
As for the Hirsfield analysis goes, I was confused since the Analyze → Properties → Atomic properties didn’t give the values of Hirsfield population, It’s in the screenshot.
I’m comparing fukui functions with all other charge populations to quantity the electrophilic sites.
I’ll look into the zeros for the Hirshfield charges in the table. That’s a clear bug.
IMHO, you’d want to use Hirshfield charges over Mulliken for comparing electrophilic sites. Mulliken charges are problematic, not least because they’re basis set dependent.
But I’d guess the Fukui indices should be the best. Incidentally, if you can share an example file, I’ll see what I can do about adding Fukui parsing to the code.
if you can share an example file, I’ll see what I can do about adding Fukui parsing to the code.
So I calculate the condensed Fukui Functions with Multiwfn, it generates a plain txt called CDFT.txt.
Hope it helps!
edit: I believe with Multiwfn, the atom indices start with 1 rather than 0 (default in orca). Apart for this difference the position are compatible with orca.
Let me think if there’s a nice way to do that in Avogadro using cclib .. and add the Fukui indices as labels. Should be pretty easy to write a script that looks for the CDFT.txt file too.
I had a glance, I believe the python script is DFT code dependent (in that case, it’s Gaussian).
I had a discussion about Fukui functions on ChimeraX mailing list, I think the info below might be useful if you plan to adding functionality within Avogadro.
As for writing the script to read CDFT.txt file, I’m interested in this!
I looked up the repository, I believe all the output processing scripts are located in avogadrolibs/avogadro/quantumio/. If I’m guessing it right, I need to write fukui.cpp and let it read CDFT.txt file. I can see this is done on orca.cpp for “MULLIKEN or LOEWDIN”.
While I agree that Mulliken charges are quite problematic (god forbid CO2 actually show the correct bond dipoles across basis sets), I’d caution against Hirshfeld charges. The original Hirshfeld method is not quite as robust as it was intended to be, and for good reason had been updated to an iterative stockholder method. That method still isn’t entirely without problem though (e.g. the bifurcation problem), and doesn’t always provide a physical description of the charge distribution (not than any charge partitioning does, but as the saying goes, all models are wrong, some models are useful).
Perhaps the gold standard of charge partitioning is the DDEC6 method, which can be finnicky to get working but generally is more robust than other methods and is not susceptible to the bifurcation problem.
I think that in many situations where Mulliken charges are still being used, simply switching to Löwdin charges would already be a meaningful improvement. Löwdin analysis keeps the same basic population-analysis philosophy, but by using a symmetry-orthogonalization is more stable.
However, as someone working mostly with plane-wave calculations, I usually don’t have access to this kind of quick-and-dirty population analysis unless I explicitly localize using LCAO basis. In that context, real-space methods feel more natural.
Bader charges seem more appropriate, since they are directly derived from the electron density and do not depend on any particular basis set. Of course, Bader analysis has its well-known limitations: it often overestimates ionicity because the zero-flux partitioning can assign disproportionately large basins to certain atoms, leading to rather extreme charge values.
This is where methods like DDEC6 become attractive. By incorporating chemical and electrostatic considerations, DDEC6 tends to produce charges that are more chemically intuitive and transferable. In that sense, it complements Bader rather than replacing it.
Overall, I feel that all of these methods have their place. Mulliken, Löwdin, and Bader are particularly valuable from an educational perspective because they are relatively easy to implement, which helps illustrate why atomic charges are not uniquely defined and how different partitioning schemes naturally lead to different results.
I have also seen some work where people report both Bader and DDEC6 charges, or even use their average, to identify more robust trends. While this is not strictly rigorous, it can sometimes provide reasonable trends.