Sorry if this has been raised before, if it is planned, or even if it’s possible to do it already and I just don’t know how.
I was wondering if it would be possible to reimplement, or extend, the bond-centric manipulation tool in such a way as to make it more intuitive, and useful? To my mind, the natural default behaviour I would expect is that when I select a bond, and then drag one of those atoms in the bond, the whole fragment attached to that atom would move as one? I.e. not just one atom moves, as now. The structure should stay the same (i.e. all other internal coordinates stay the same) and only the selected bond length/angle should change, whereas at the moment moving an atom in this way has knock-on effects for other internal parameters.
Selecting a part of the molecule and using the normal manipulate tool moves the whole selected part as one, but relative to the xyz frame (also very useful!) rather than a bond.
Equally, it would be extremely useful to have a way to manually adjust dihedral angles/rotate whole substituents around a bond. The tool should do what we as chemists think of as bond rotation, i.e. rotation of a whole fragment around a bond. It should be possible to easily and intuitively adjust the rotation of a methyl group, or of the two rings in a biphenyl system, etc. Perhaps by choosing a bond using the tool, then clicking and dragging an adjacent atom (rather than a member of the bond).
Being able to manually adjust geometries around the internal coordinates in this way would be very helpful for converging an structure to a minimum, for example, as you often want to just slightly rotate a substituent or slightly lengthen a bond, if they are geometry changes associated with a remaining negative frequency. At the moment this doesn’t seem to be simple…
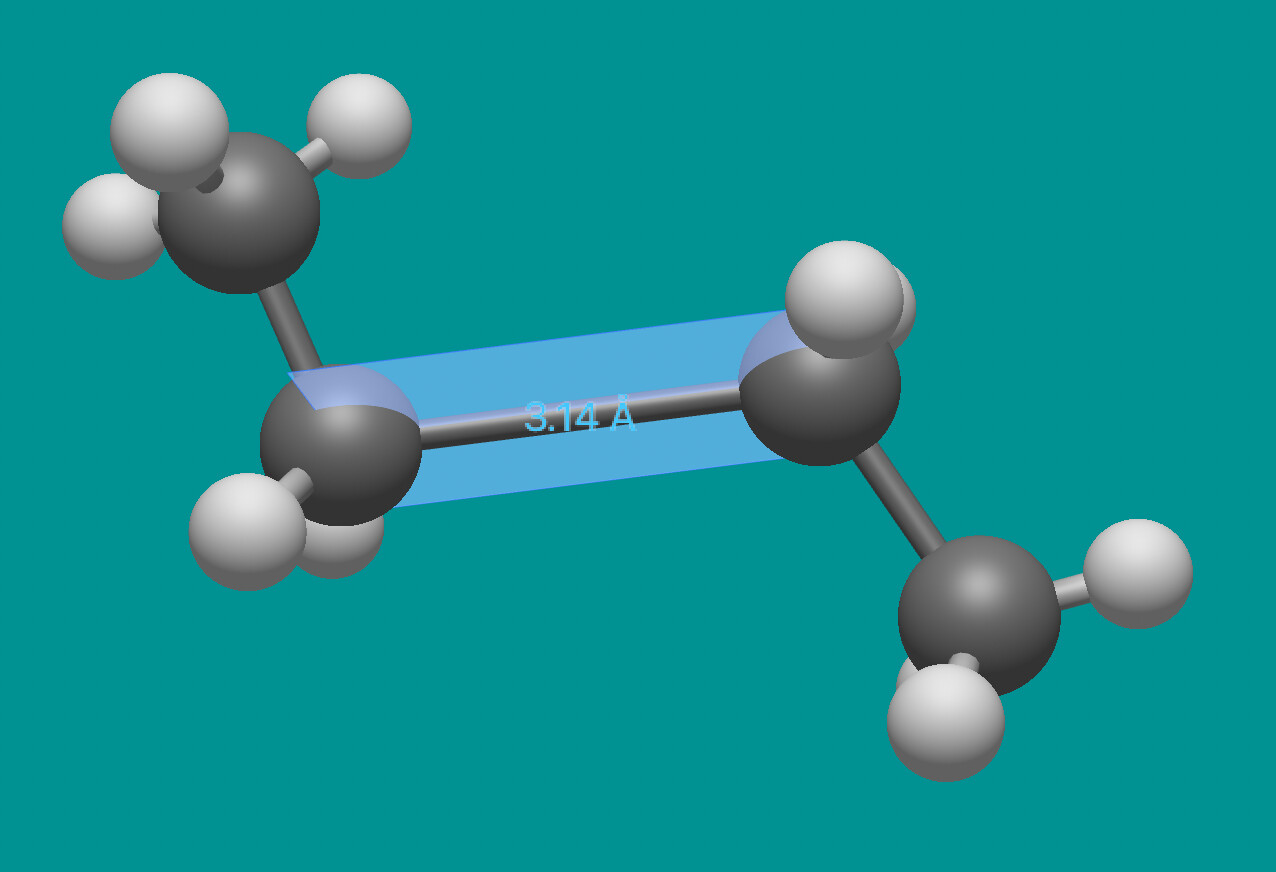
You click on a particular bond and then right-click or control-click on one of the atoms and the whole connected chunk will move (e.g., stretching a bond to 3.14Å)
In v1.x you could also do this through the Bond Property, Angle Property, Torsion Properties tables (e.g., type a specific value). That’s not yet supported but basically, I’m re-using the same code from the bond-centric manipulation tool.
It sounds like you’re either using the manipulate tool (i.e., atoms and XYZ chunks) or there’s a bug.
Ah, so I just tested various situations again. Indeed, for the butane example it also works as you describe and as I would expect. Which is possibly why I never noticed the problem until a couple of days ago.
However, it seems that if one of the atoms in the bond is a member of a ring (regardless of whether the ring is sp2 or sp3) then it stops working as behaved, and only the atom that is clicked can be moved - nothing else moves with it. A ring behaves as expected if it is attached to one of the atoms in the selected bond, only not if it contains the atom in the bond. The easiest example to test for yourself is a biphenyl system I suppose. Trying to change the Ph-Ph bond length or the angle between the rings highlights the problem.
Perfect, thanks for filing the bug report! Though note that it is not restricted to biphenyl systems, it happens with any atom within any ring, in case I wasn’t clear
Manipulating ring bonds is very tricky. That’s why when you move a “ring bond” it should only move that atom. In this case, the definition of “ring bond” is wrong. It’s flagging a “ring bond” when it’s simply between two atoms in rings.
Otherwise, I’m clear that it’s not just biphenyl… That just happens to be a very clear test case.