I would like to express my sincere gratitude for your hard work and dedication to developing Avogadro2.
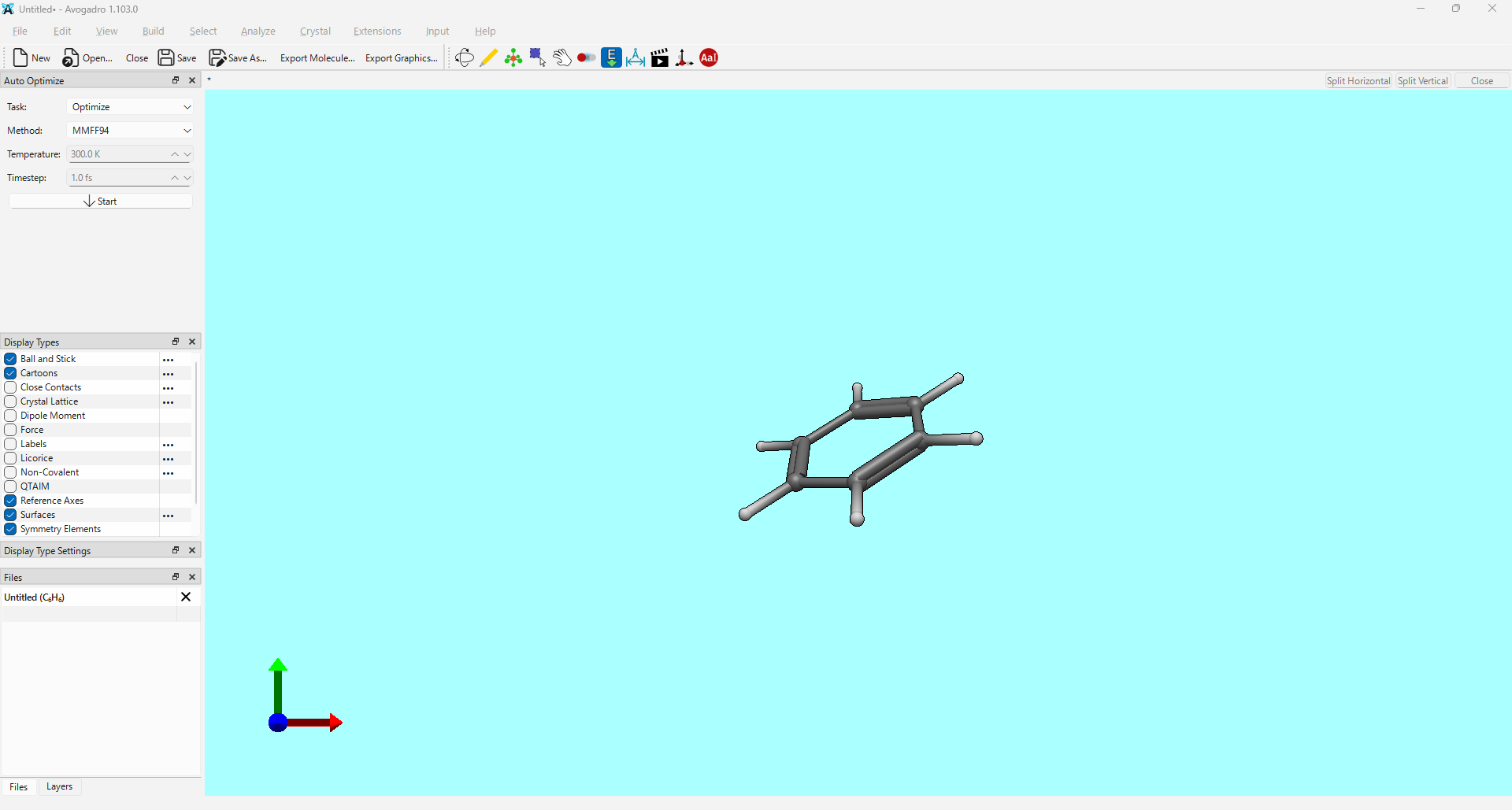
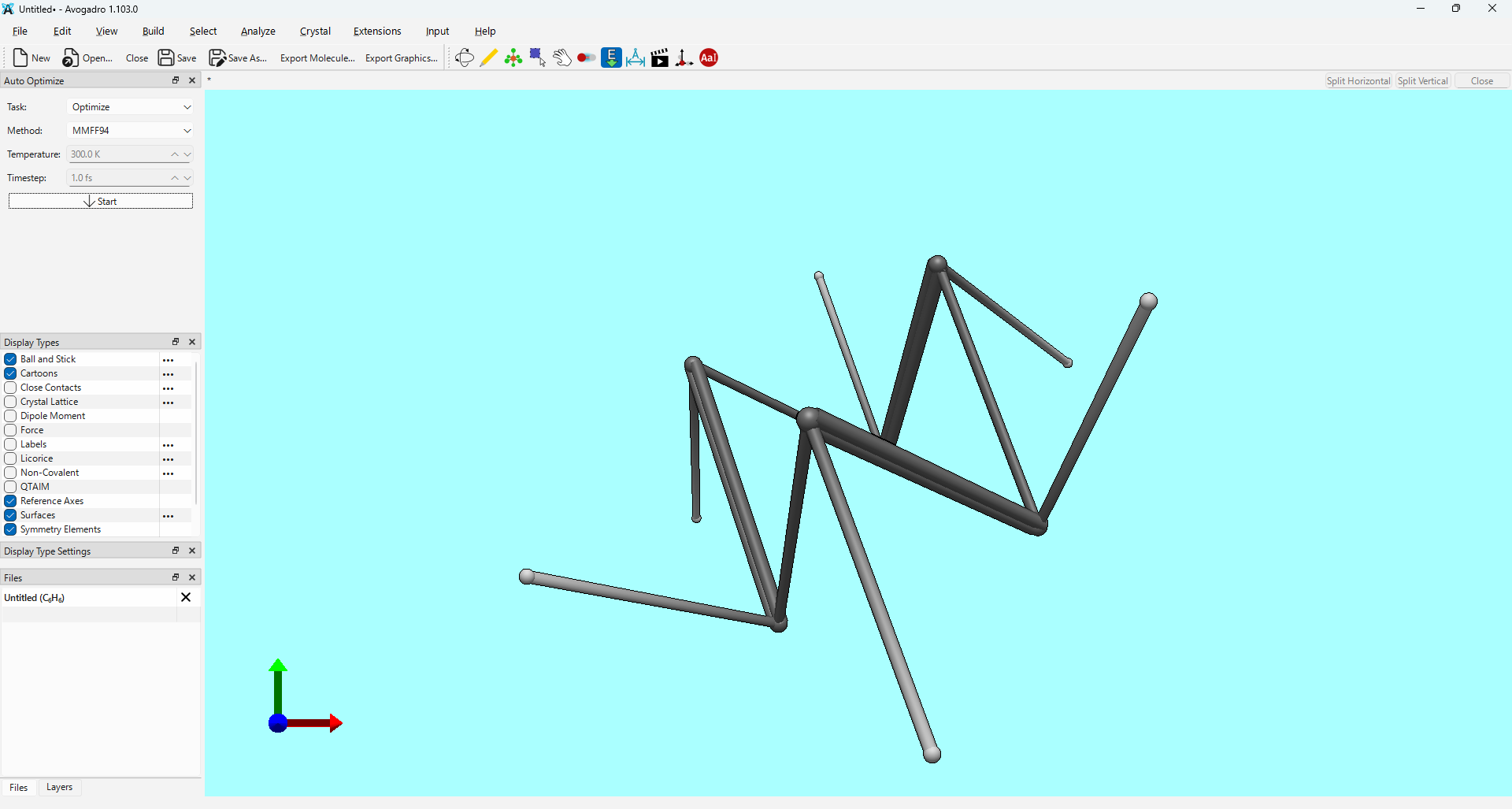
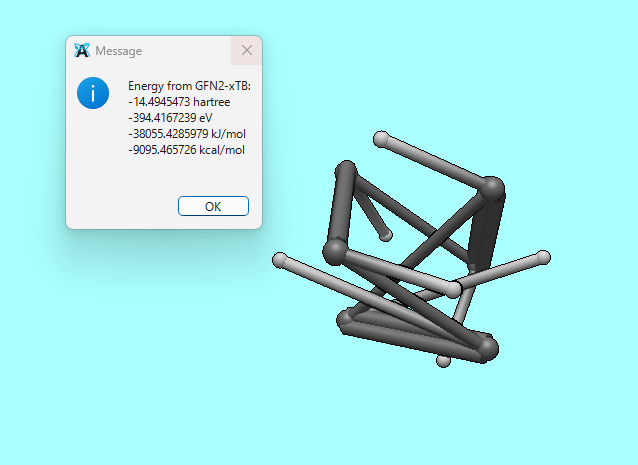
I’ve noticed an issue when performing geometry optimization using the xtb extension within Avogadro2. As shown in the attached screenshot, the molecular structure appears to distort significantly during the process.
This issue occurs in the latest nightly build (version 1.103.0 for windows11), which I downloaded on March 30, 2026.
I used the following settings for the optimization:
Optimization Level: vtight
Solvation: none
Method: GFN2-xTB
Notably, the same distortion occurs regardless of the method; I have confirmed that GFN1-xTB and GFN0-xTB also lead to the same result.
Yes, I am using the xtb plugin. To be specific, I run Extensions > Semi-Empirical QM (xtb) > Optimize first, and after that, I execute Extensions > Semi-Empirical QM (xtb) > Energy.
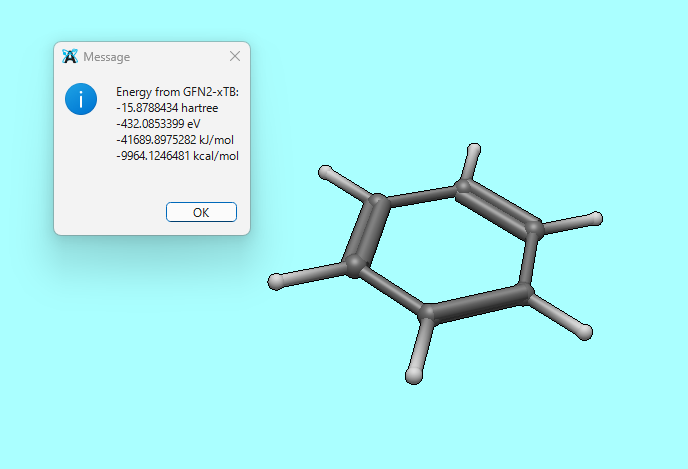
Just for comparison, the following is the result of running Extensions > Semi-Empirical QM (xtb) > Energy after performing an initial geometry optimization with MMFF94.
I was able to reproduce this myself on Windows, and I think I’ve identified the problem to be an xTB/conda bug, not a problem with the xTB plugin.
It seems that, for some reason, the Conda version of xTB is not functioning properly on Windows (which has been noted by others), but the actual precompiled binaries available from the xTB GitHub page do function properly. Thankfully the xTB plugin makes it pretty trivial to switch which xTB executable you’re using. Just go to Extensions ⇒ Semi-Empirical QM (xTB) ⇒ Configure... and, after you’ve downloaded the Windows binaries from the GitHub page, select xtb.exe as your binary.