Hello! I am trying to generate conformations and get their energy values for some molecules.
I recalled in the previous versions, this function was just there… but I am using Avogadro2 and it seems like it’s not there anymore? Should I be installing something?
As a bypass, you can instruct openbabel (from the CLI) for this task. See the manual here and here for the parameters available. To provide an example about diethyl ether, enter the two commands
which first generates a starting point, then probes for up to 50 conformers. Note, --writeconformers ensures all conformers identified enter the permanent record written; else, there would be only one single entry.
I don’t know how to display them in Avogadro one by one as e.g. in Jmol, or all simultaneously and centred in one visualization in common (perhaps in different colors for an easier discern), however it definitively is possible to split the set by a subsequent
obabel conformers.xyz -O single_conformer.xyz -m
into multiple data files each with one conformer only.
Yeah, someone pointed this out a bit ago - I didn’t notice because my group generates conformers using obabel either on the command-line (as @Thomas indicated) or in our Python scripts.
Thank you! May I know how do I display my results after typing into the command line?
My goal is to find out how many possible conformers there are, and identify which is the lowest energy conformer. How do I do that via the command line?
which instructs openbabel to generate up to 50 conformers and store the results in permanent record (here: conformers.xyz, equally see the documentation here). The more bonds offer rotational flexibility (alkyl chains, non cyclic ethers, etc), the higher the number of permitted thresholds (--nconf) should be set. Note the options --random, --systematic, --weighted, etc – they can yield different conformers. You can then access the forcefield energy as a listing (manual) by
obabel out.xyz -otxt --energy --append "Energy"
in sequence of the models in you (container) .xyz file.
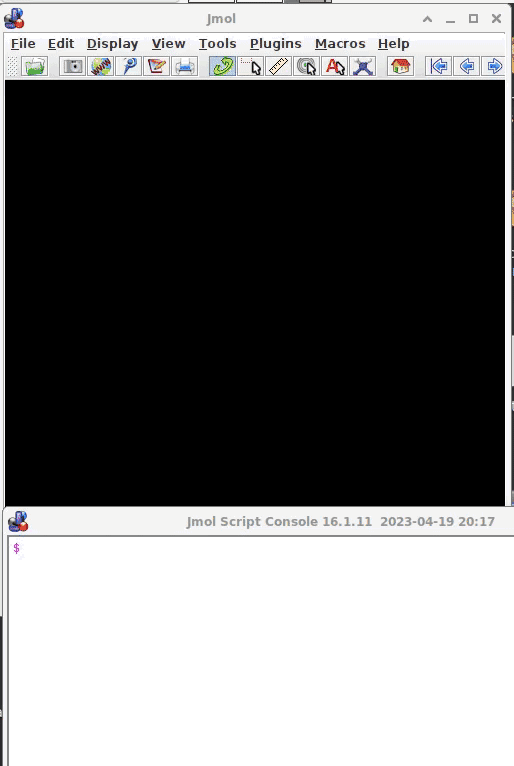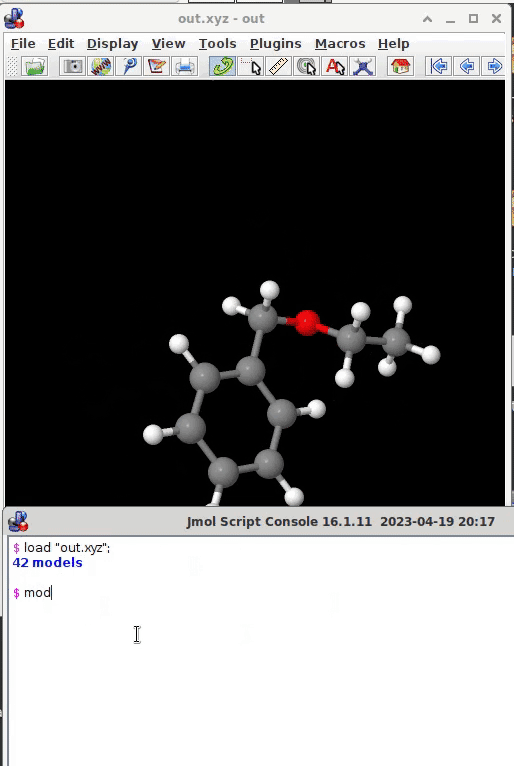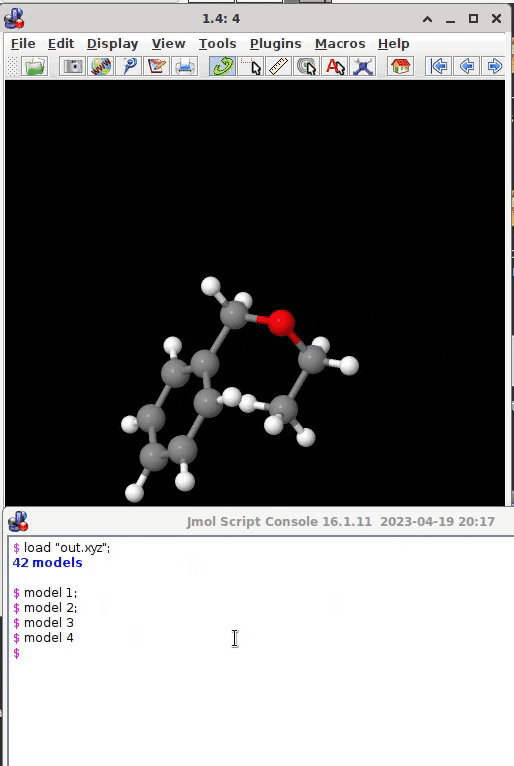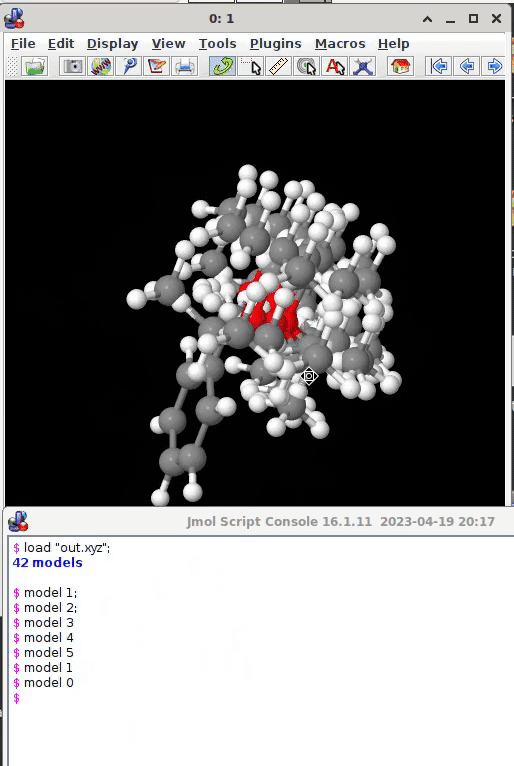
As for the display of the individual conformers, I recommend Jmol.* Start the program in the folder with the data to visualize (some cd to enter the correct location might be necessary), which has its own command line interface (File → Console) and issue
load "out.xyz";
as the file containing the structure(s) of interest. The program’s console will indicate how many models the .xyz file contains, the canvas displays the very first one model. You then can can manually pick one after the other by
model 1;
model 2;
model 3;
or all models present at once (by model 0). The wikipage and the interactive documentation provide much additional information what Jmol can offer (an ocean of video tutorials on youtube, too):
* Maybe there is a way to display/to browse through multiple conformers using Avogadro (perhaps with an indicator of the e.g. UFF force field energy computed. So far, I’m simply not aware if this is a feature already implemented in Avogadro/or in preparation for a future edition.
@jenmong There should be multiple conformers. However, it depends both on the number of single bonds with free rotation as well as the parameters engaged to identify conformers (systematic, weighted, etc) if the -nconf 50 really leads to 50 entries. In the case of benzyl ethyl ether below, there criteria were met for 47, i.e. there could have been three additional entries to report. On the other hand, the search would have been incomplete for a query of e.g., n-octane with many more degrees of freedom.
For the file below: remove the file extension .txt to access a .zip archive. Decompress the .zip archive. Then file my_log.md describes an example search for conformers.
@ghutchis What is your opinion to add .zip to the file formats permitted as an attachment here? The moderated user forum of datawarrior for example set a constraint of up to 5 files for a total of 2048KB; because this volume usually is more than enough there, I think its equally good here.
I’ve favored .tar.gz or .tar.bz2 (or just .tar) as better options. (I can recall some virus issues with ZIP, but maybe that was just general Windows virus issues.)