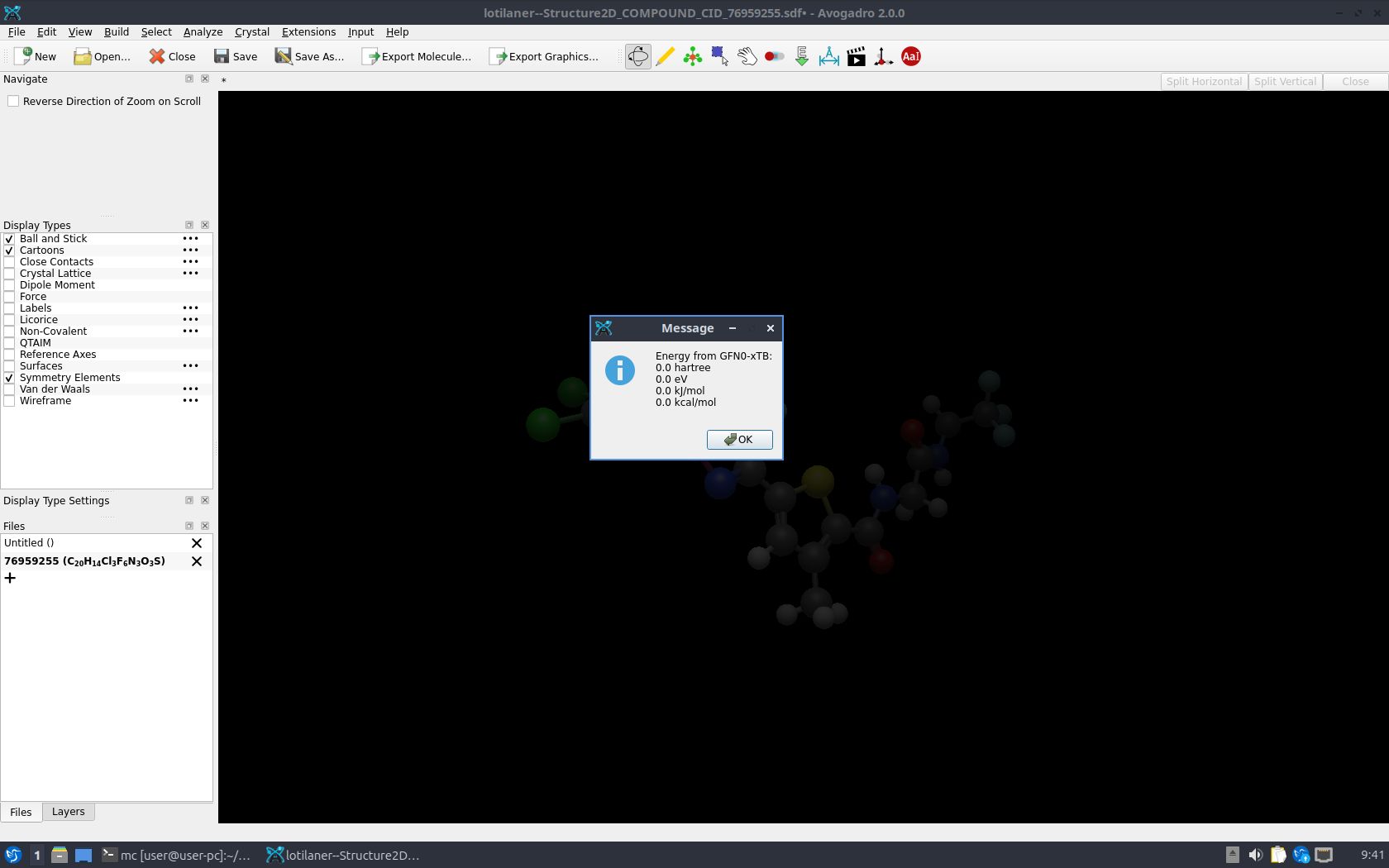
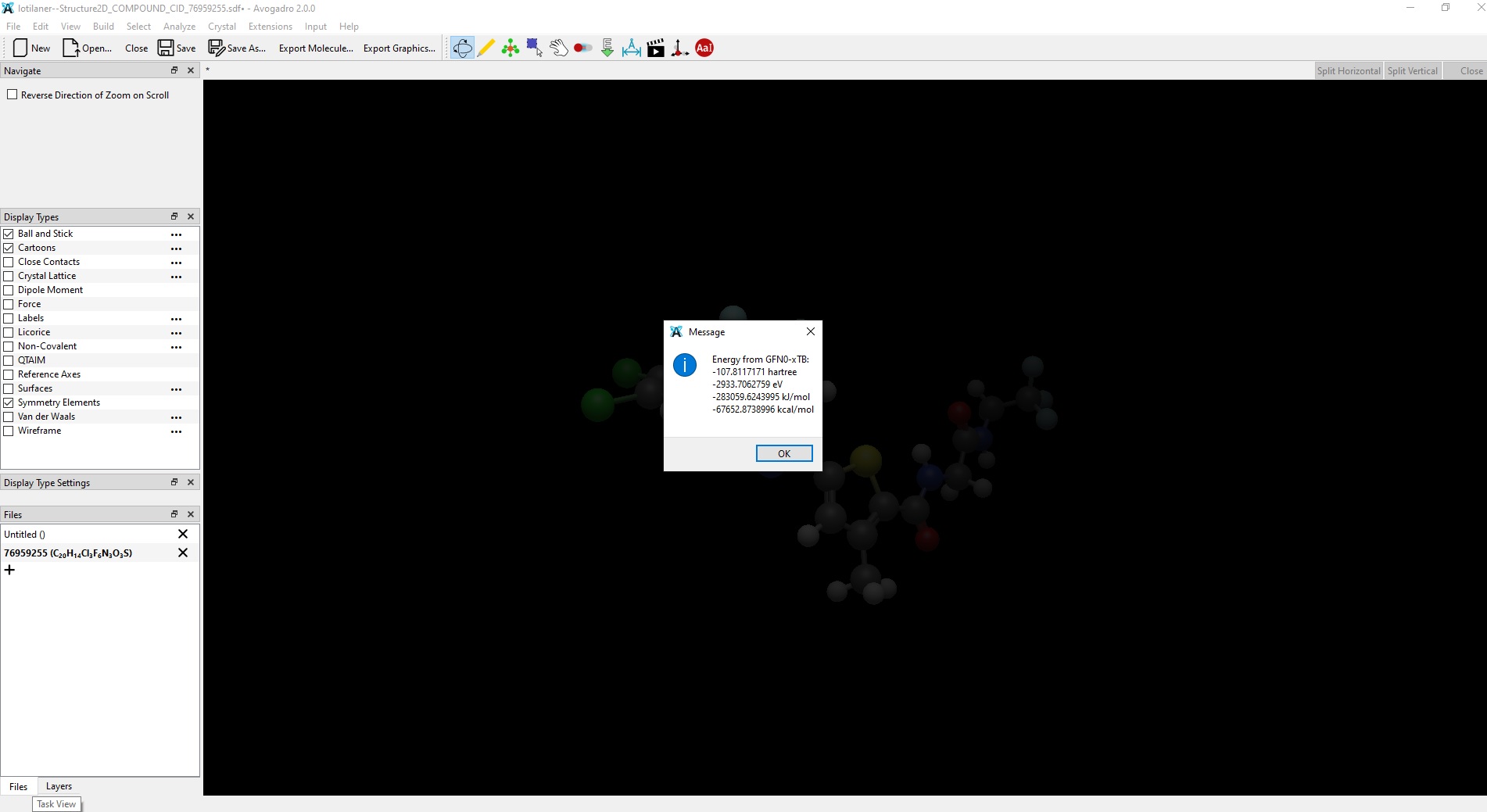
I installed the latest stable Avogadro release (2.0.0) on a Windows 10 PC and a Linux (Lubuntu 26.04, AppImage) machine. I also installed the avo_xtb (0.11.1) plugin via the Manage plugins…command in the menu of the program. When I attempt to calculate the GFN0-xTB energy of a molecule in the Windows 10 release of Avogadro, I get a reasonable (non-zero) value. The same calculation in the Linux release produces 0 kcal/mol for the energy shown in a pop-up window in the program. In the output file of avo_xtb there is the following message:
** On entry to DSYGVD parameter number 11 had an illegal value
########################################################################
[ERROR] Program stopped due to fatal error
-3- Single point calculation terminated
-2- xtb_calculator_singlepoint: Electronic structure method terminated
-1- peeq: Diagonalization of Hamiltonian failed
########################################################################
There are also some other issues with this plugin and, sadly, the Windows 10 release seems also be affected. For example, the GFN2-xTB geometry optimization of a molecule in the Linux release converges to a reasonable geometry, while the same calculation with the same molecule and the same initial geometry yields a nonsense geometry in the Windows 10 release (the energy grows in subsequent optimization cycles). In this case, there is no error messages in the corresponding output file.
I also tried a Nightly build (downloaded a week ago) but it did not help.
In general the Windows version of the plugin is not really working properly, because the version of xtb itself on conda-forge for Windows has issues. There will be a new version of the plugin at some point that gets xtb directly from the Grimme group’s GitHub repository instead of from conda-forge. I’ve started work on it, I just haven’t got round to finalizing it, sorry. I’ll try to get round to it soon.
The error you get on Linux is interesting though. I would expect that that is a bug or crash in xtb itself. Could you upload the various files?
Another issue was a geometry optimization at the GFN2-xTB level for the same initial geometry. In this case, the Linux release seems to work fine. Below my output files for the optimization of my_molecule.sdf linux-opt-gfn2-xtb.out (111.8 KB) windows-opt-gfn2-xtb.out (940.1 KB)
In the Windows release the optimization does not converge and the energy is rising (I mean less & less negative).
Yesterday I downloaded the Nightly build of Avogadro for Linux (AppImage) and installed the avo_xtb plugin as well. I carried out several tests with the GFN0-xTB method and the results seem to be correct now. Thank you for your help with this issue.
BTW, I’ve just found another issue with the avo_xtb plugin for Avogadro on Linux. The assignment of spinorbitals as the HOMO and LUMO for a biradical O2 molecule seems incorrect. The standalone version of the XTB program (on a Linux machine) yields the correct assignment. I guess this issue is also related to the conda release of the plugin. output-incorrect.out (12.0 KB) o2.xyz (138 Bytes) output-correct.out (11.9 KB)
Nothing has changed about the plugin since you last tried, though. I need to resolve a probable issue in Avogadro itself before I can release a new version of the plugin.
I’ll have a look at your other files when I get the chance, thanks. The log that the plugin produces would be very helpful as well, it should be in ~/.local/state/easyxtb/, but note that it gets overwritten with every calculation, so it needs to be the log file from just after the calculation has completed.