I recently had a discussion with another computational chemist, and they mentioned that it is possible to use varying isosurface values to “fudge” the numbers a bit, per se, with regards to showing molecular orbital overlap in molecules.
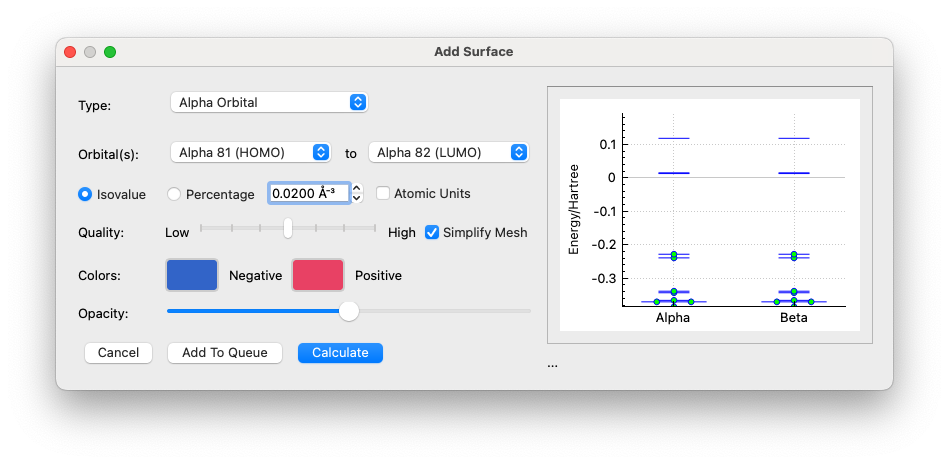
There are issues with setting a single value of course, as large molecules and molecules with large atomic number elements will have drastically different requirements for isosurface values. The current default in the surfaces dialog is 0.03, which is generally okay, but the current default in the molecular orbital window is 0.01, which is, for most practical cases, way to diffuse. I’d say that at a bare minimum they should both be 0.03, however I think other values should be considered. The values that were suggested were 0.08 and 0.0266, however I think that there is some wiggle room and some debate to be had about the merits of different values.
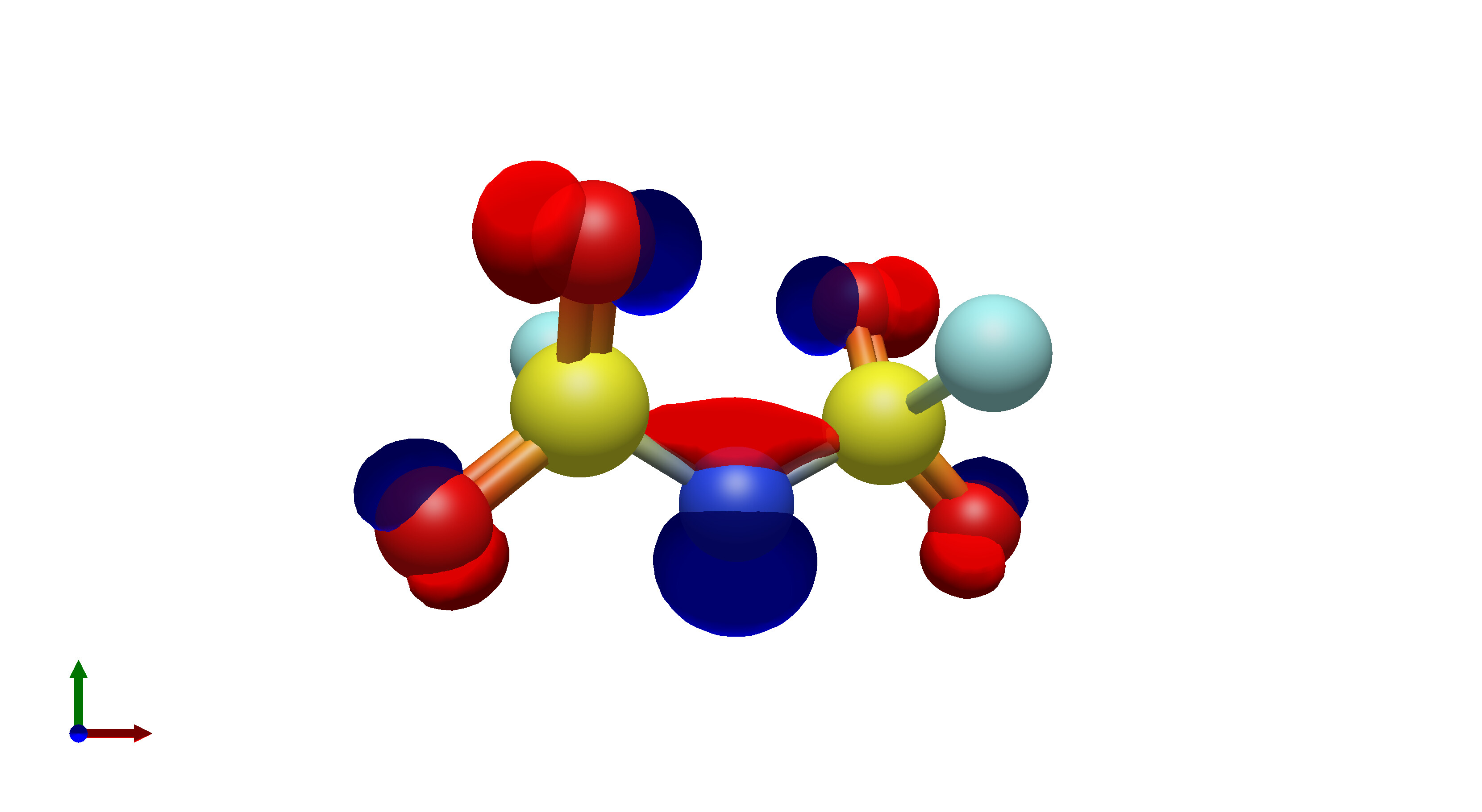
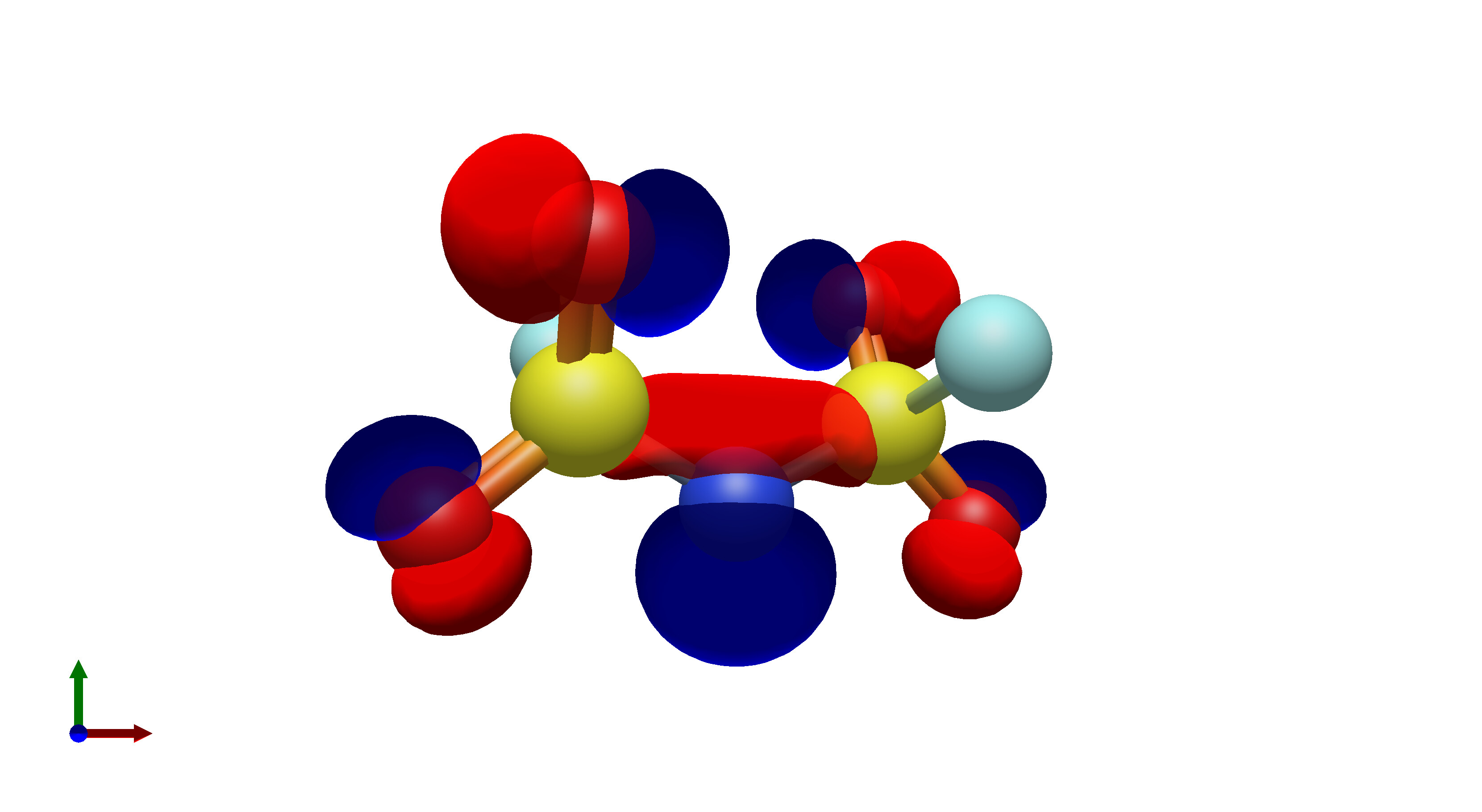
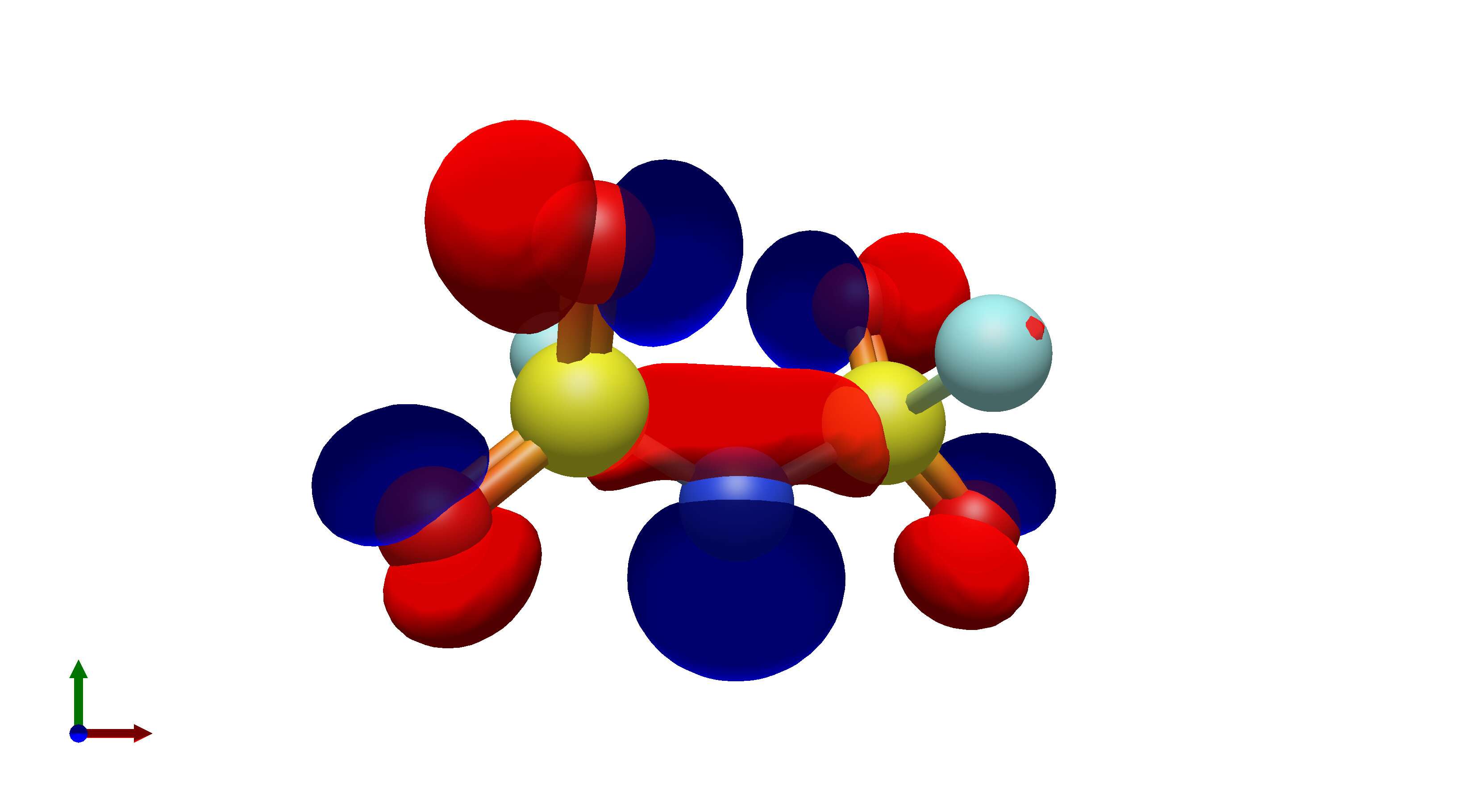
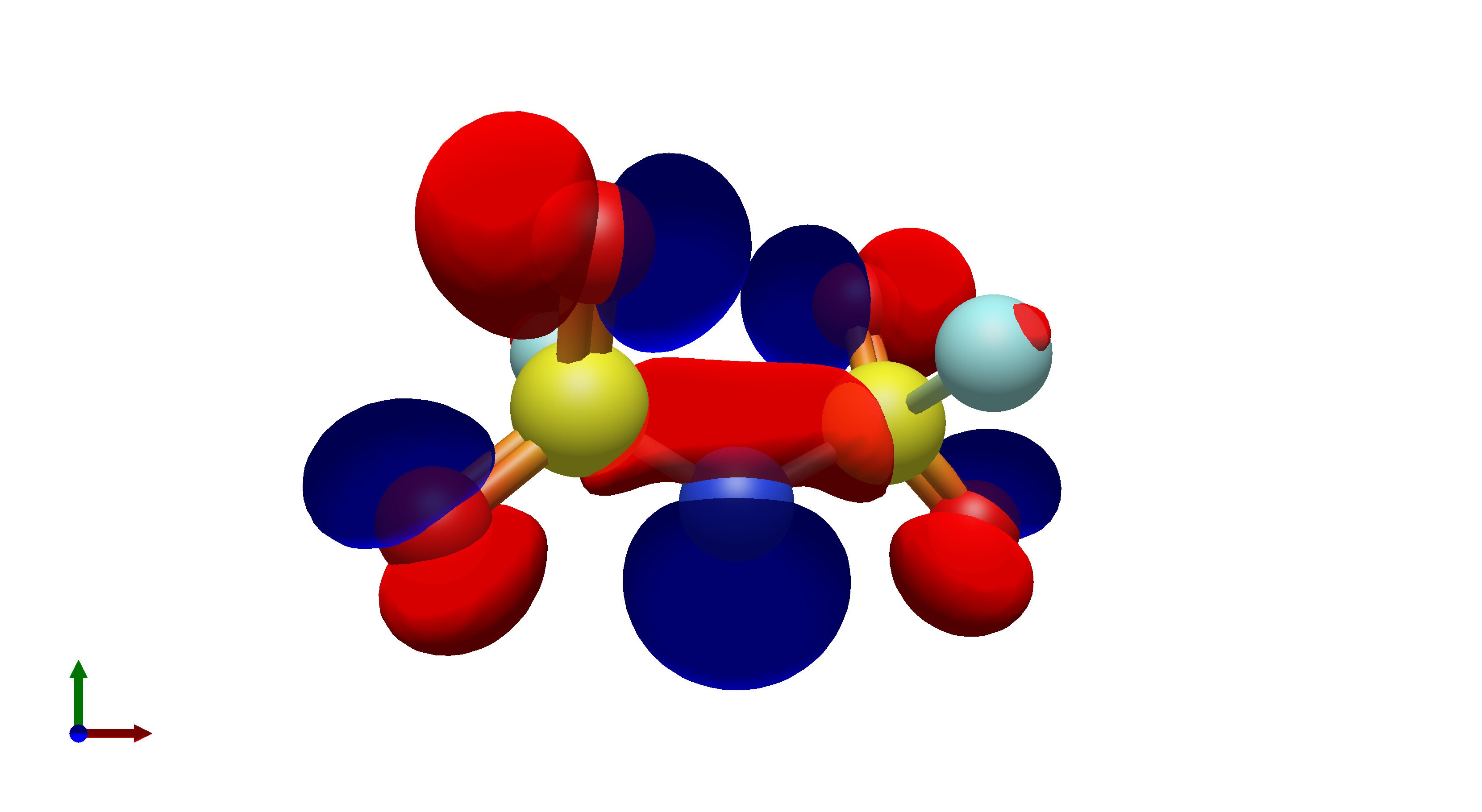
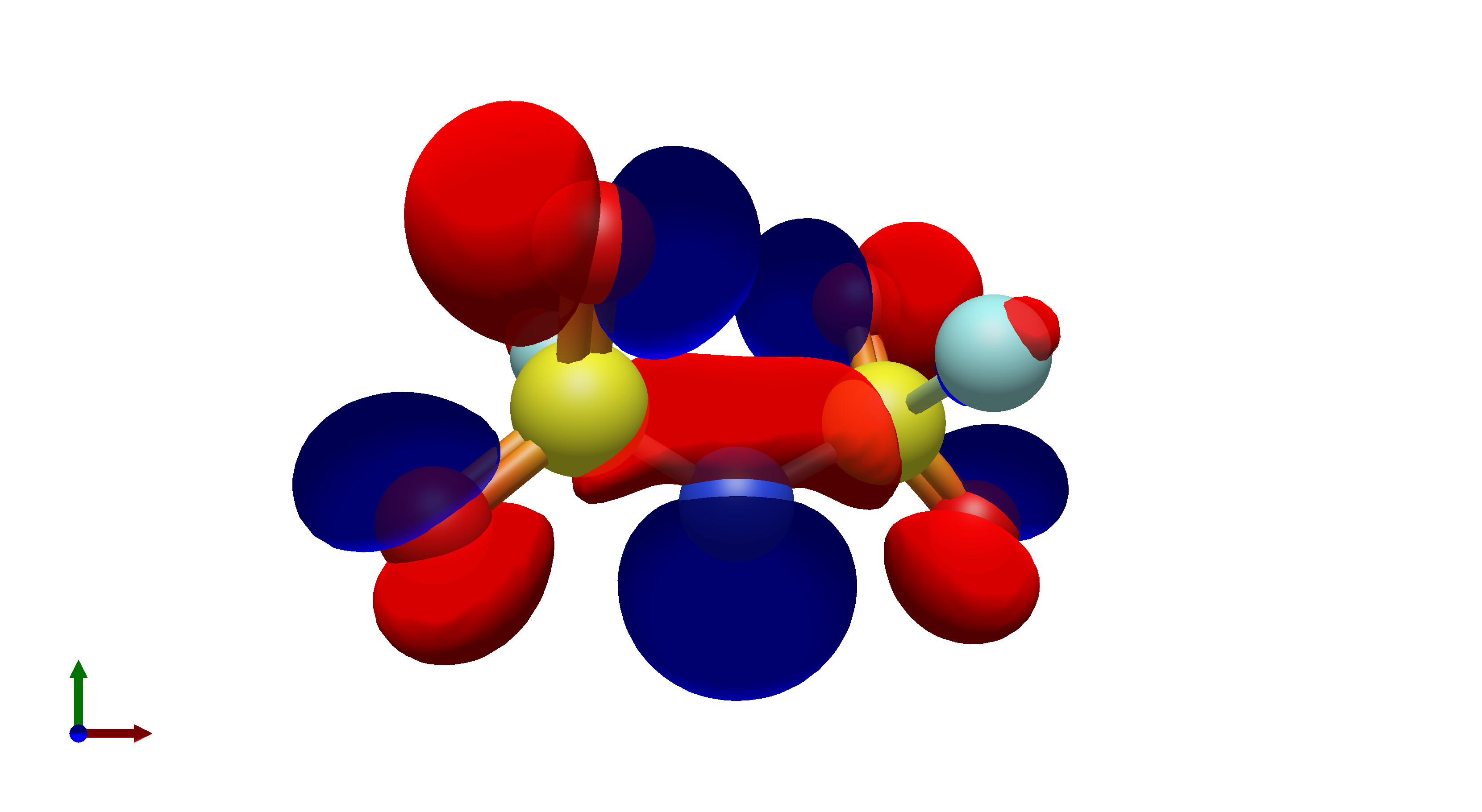
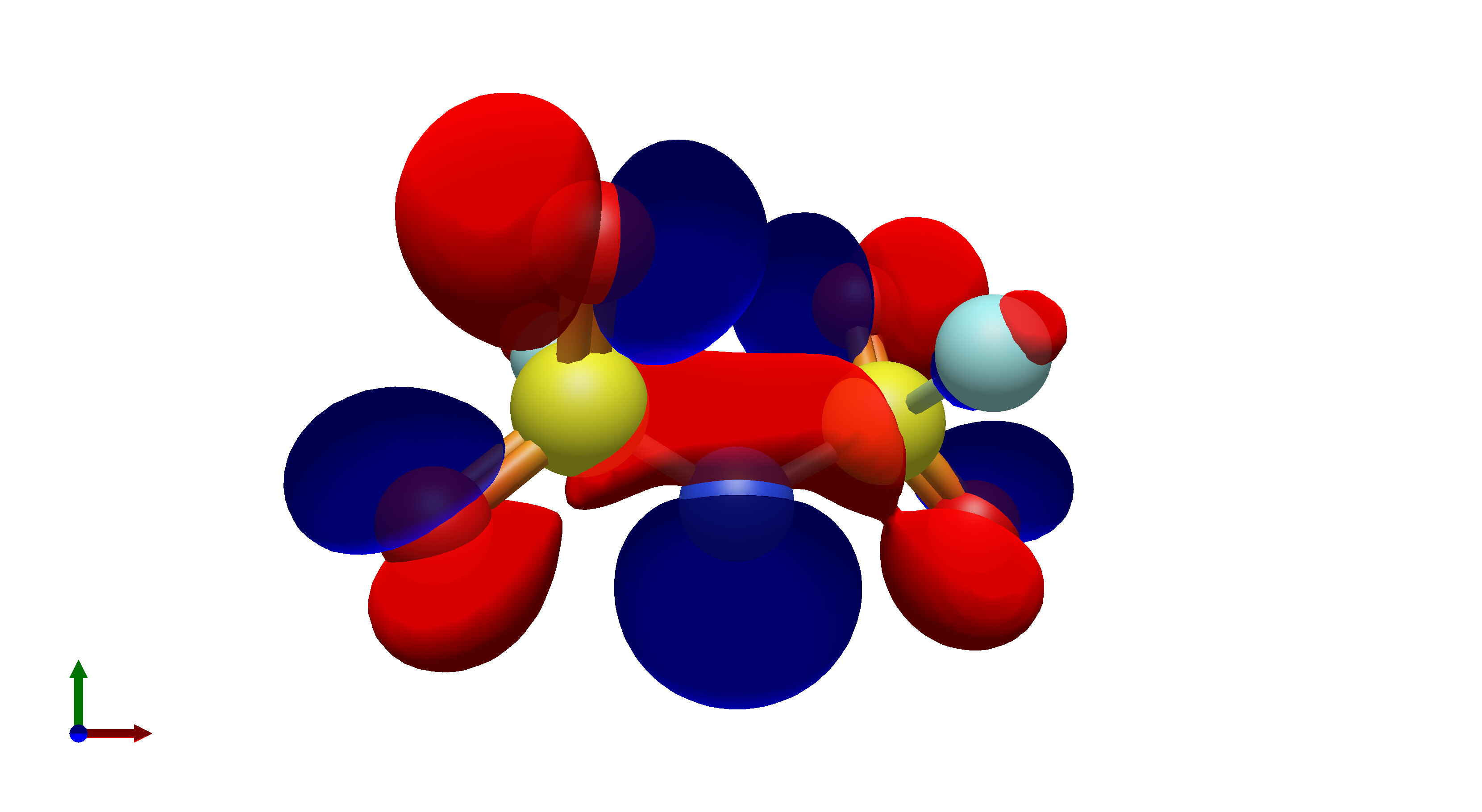
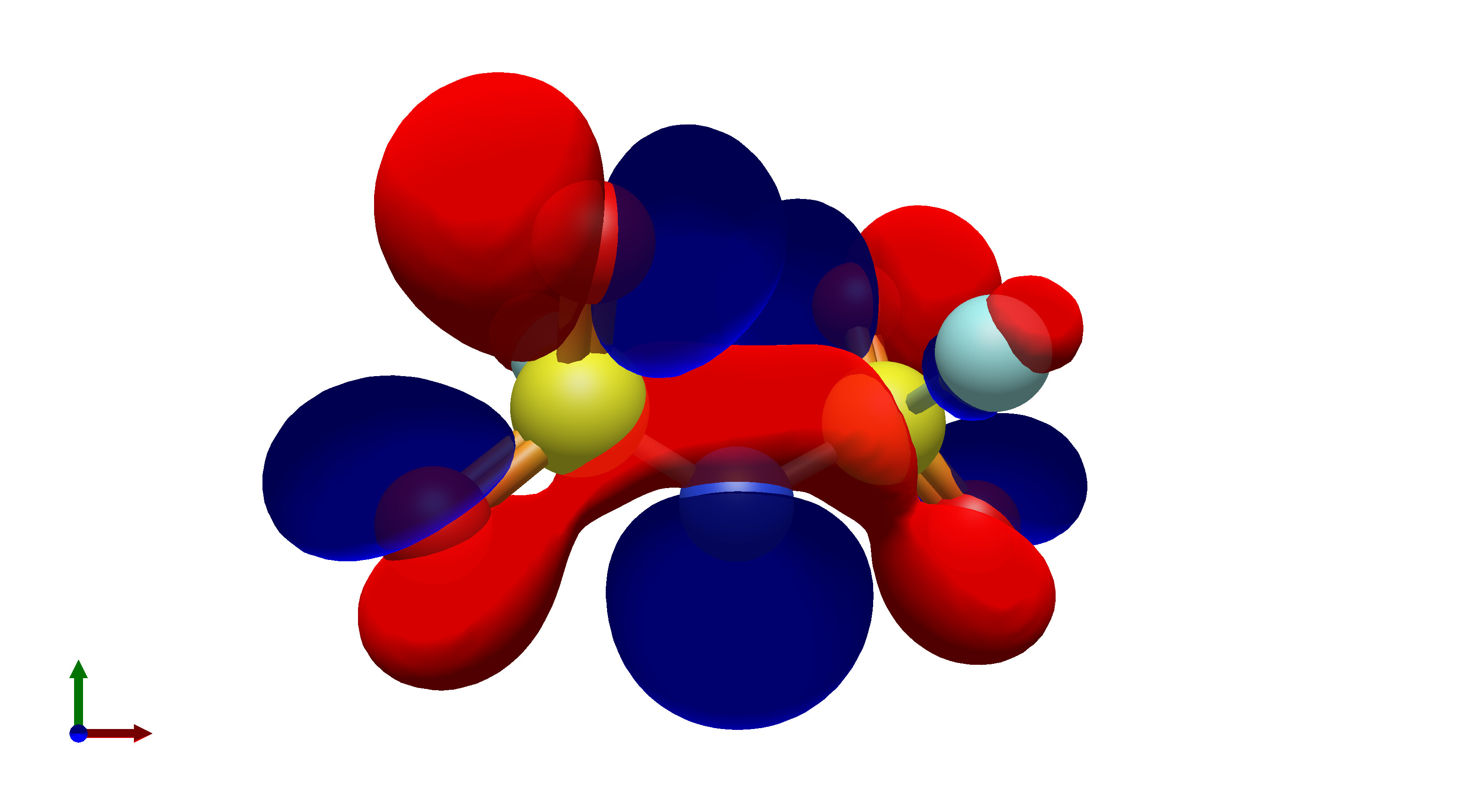
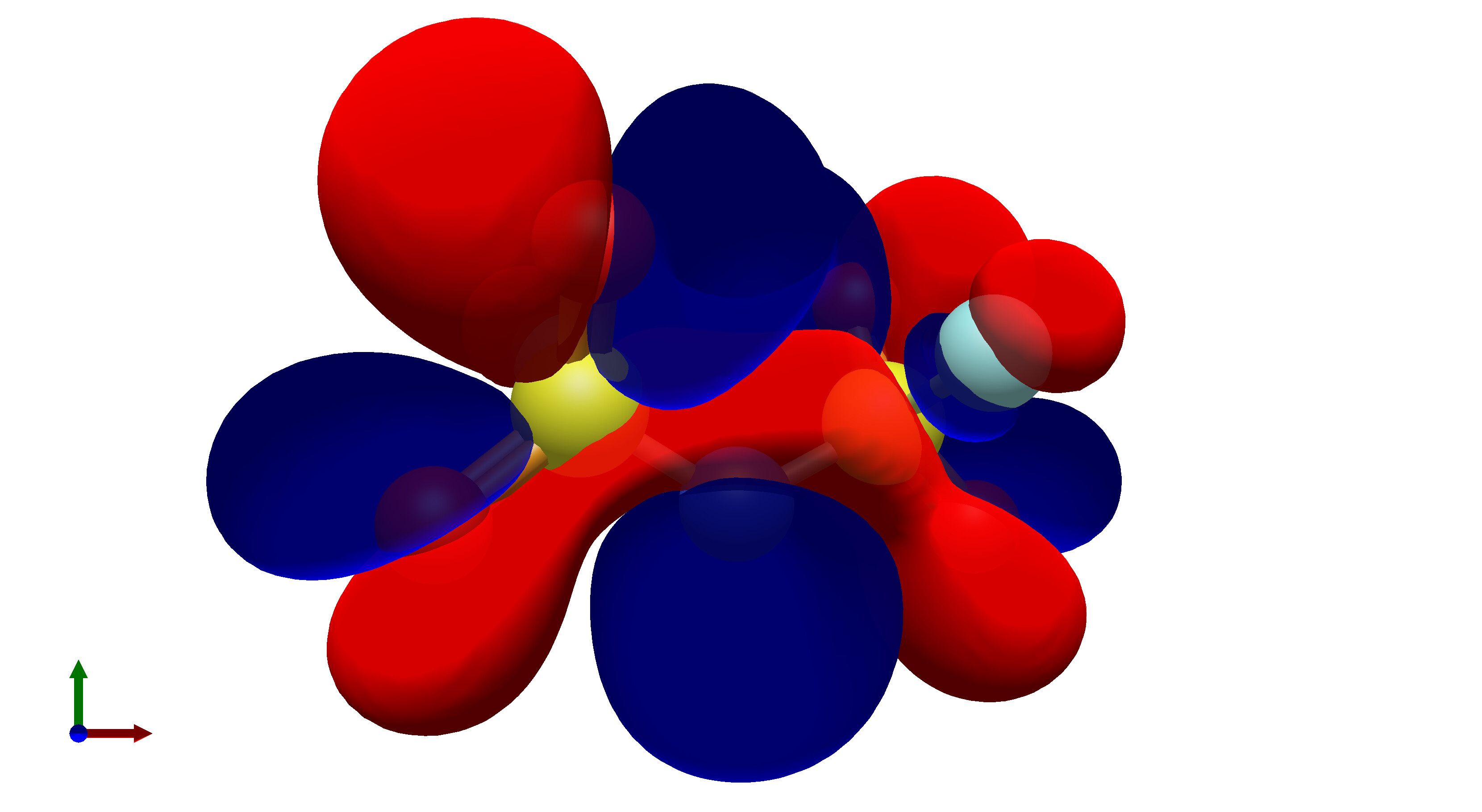
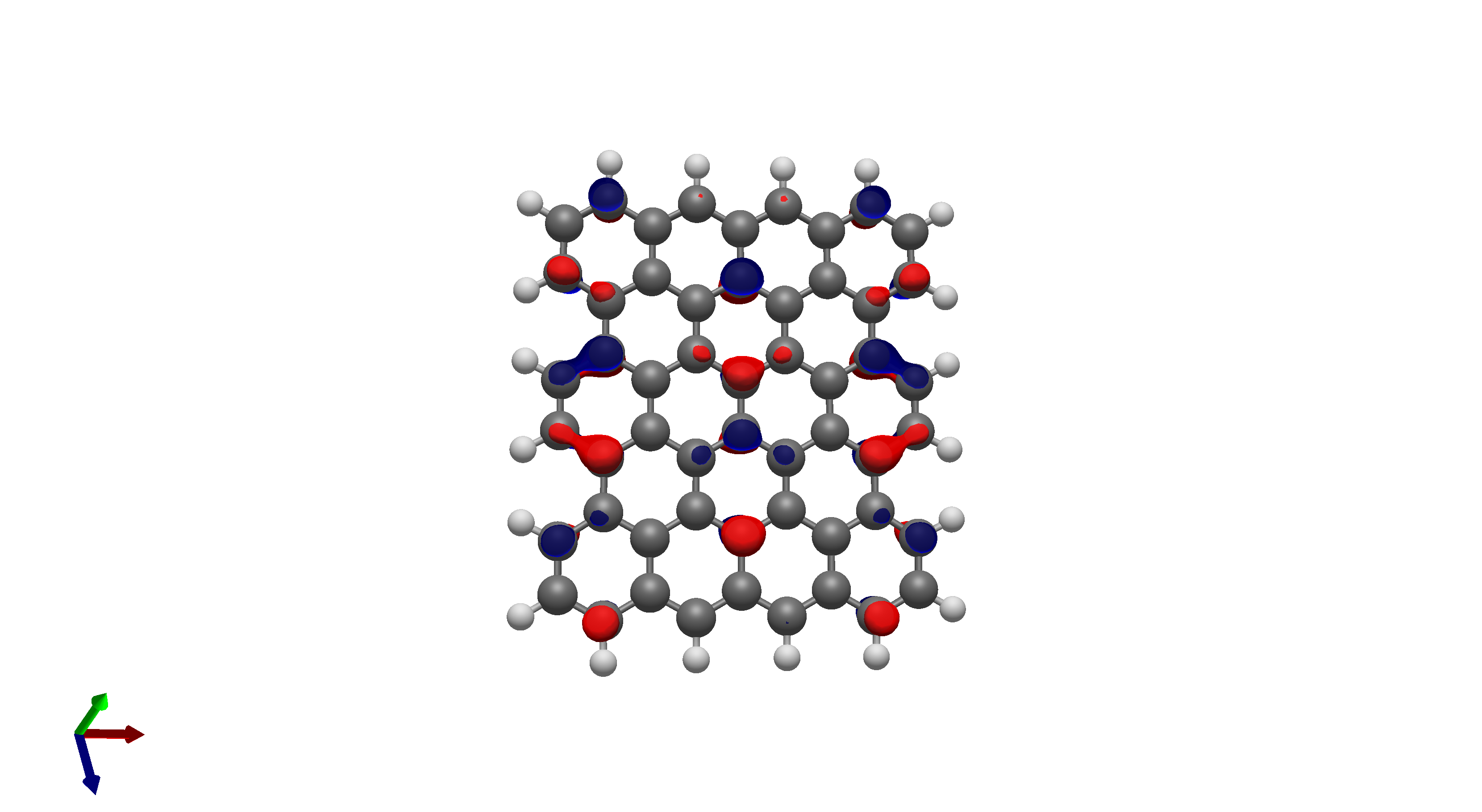
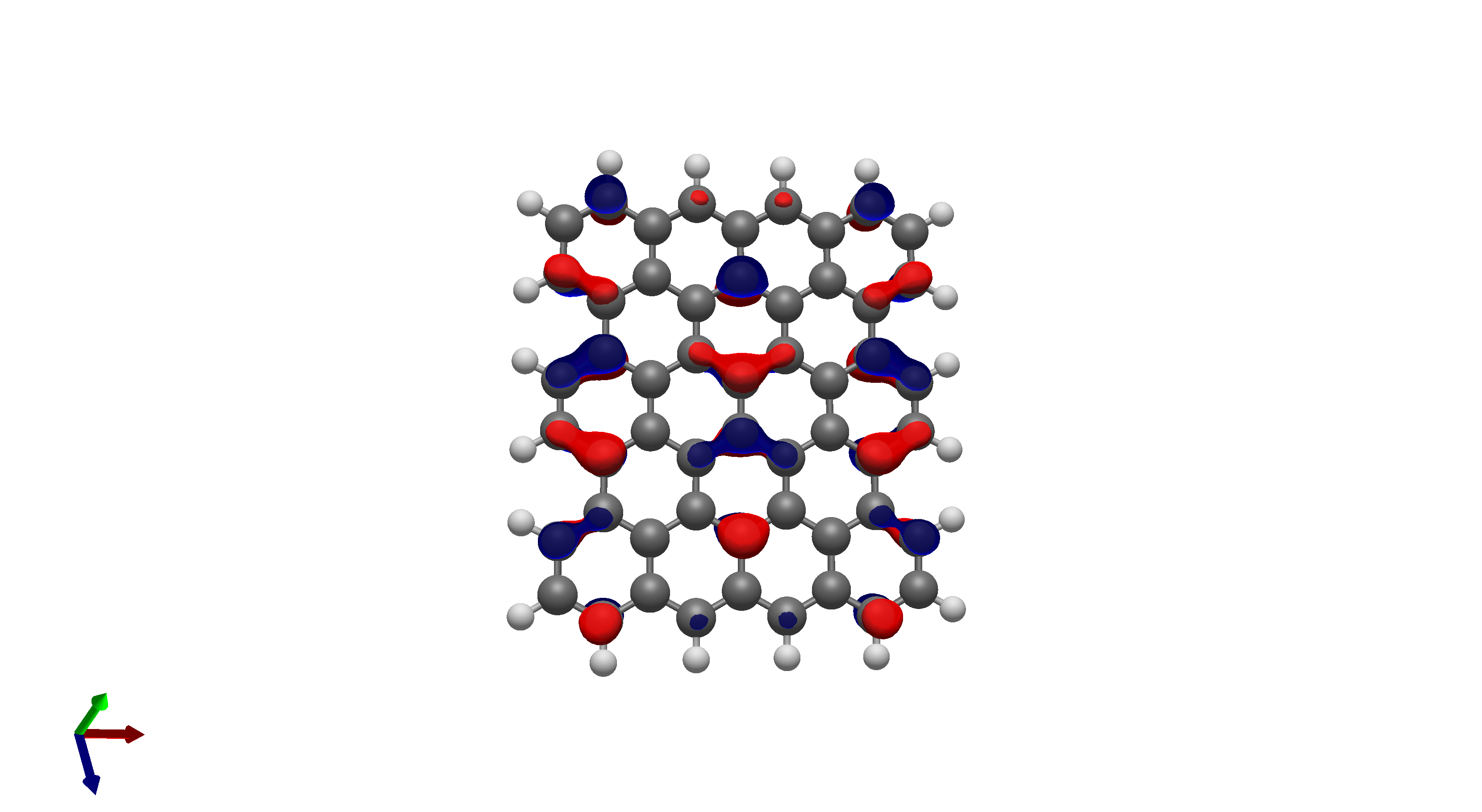
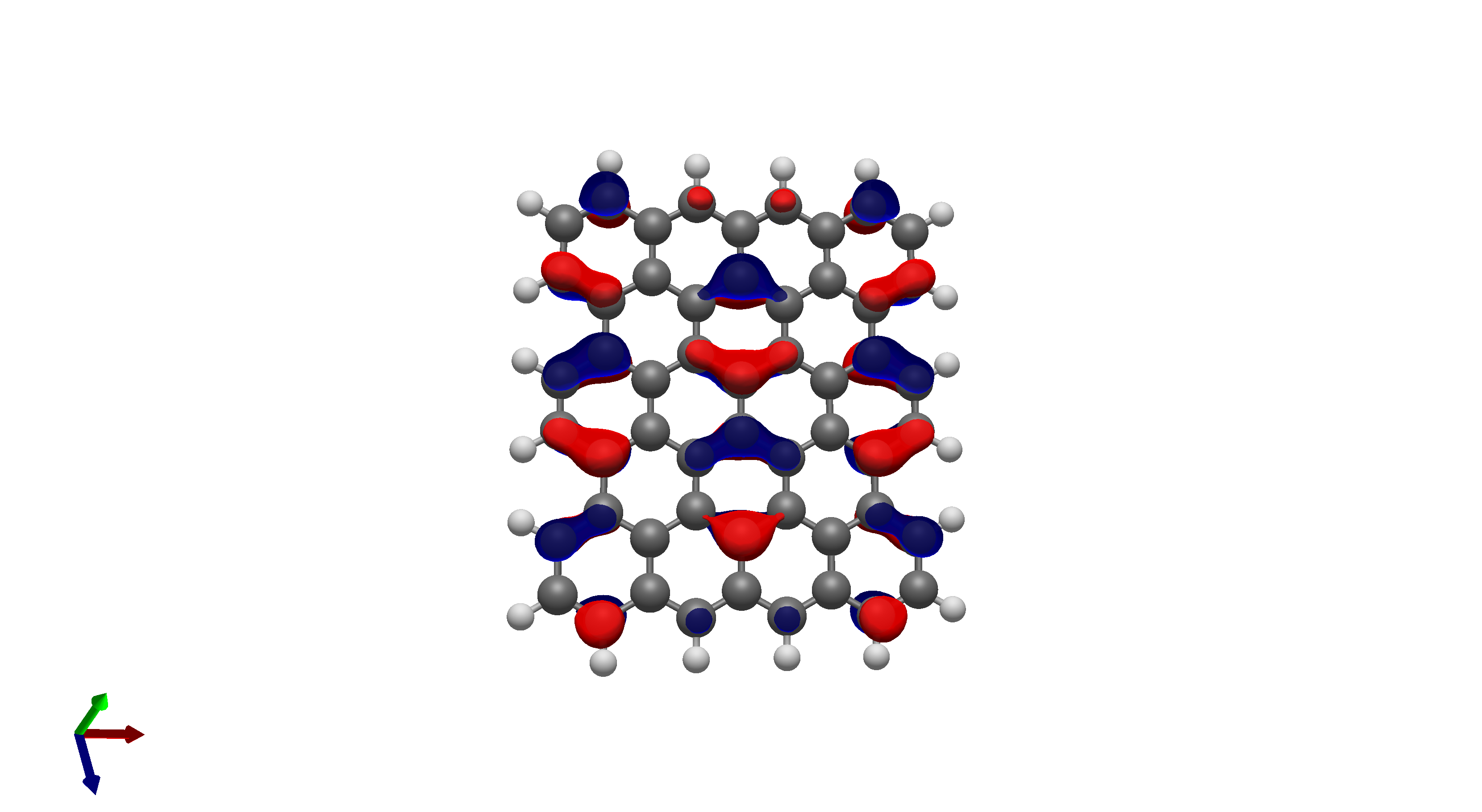
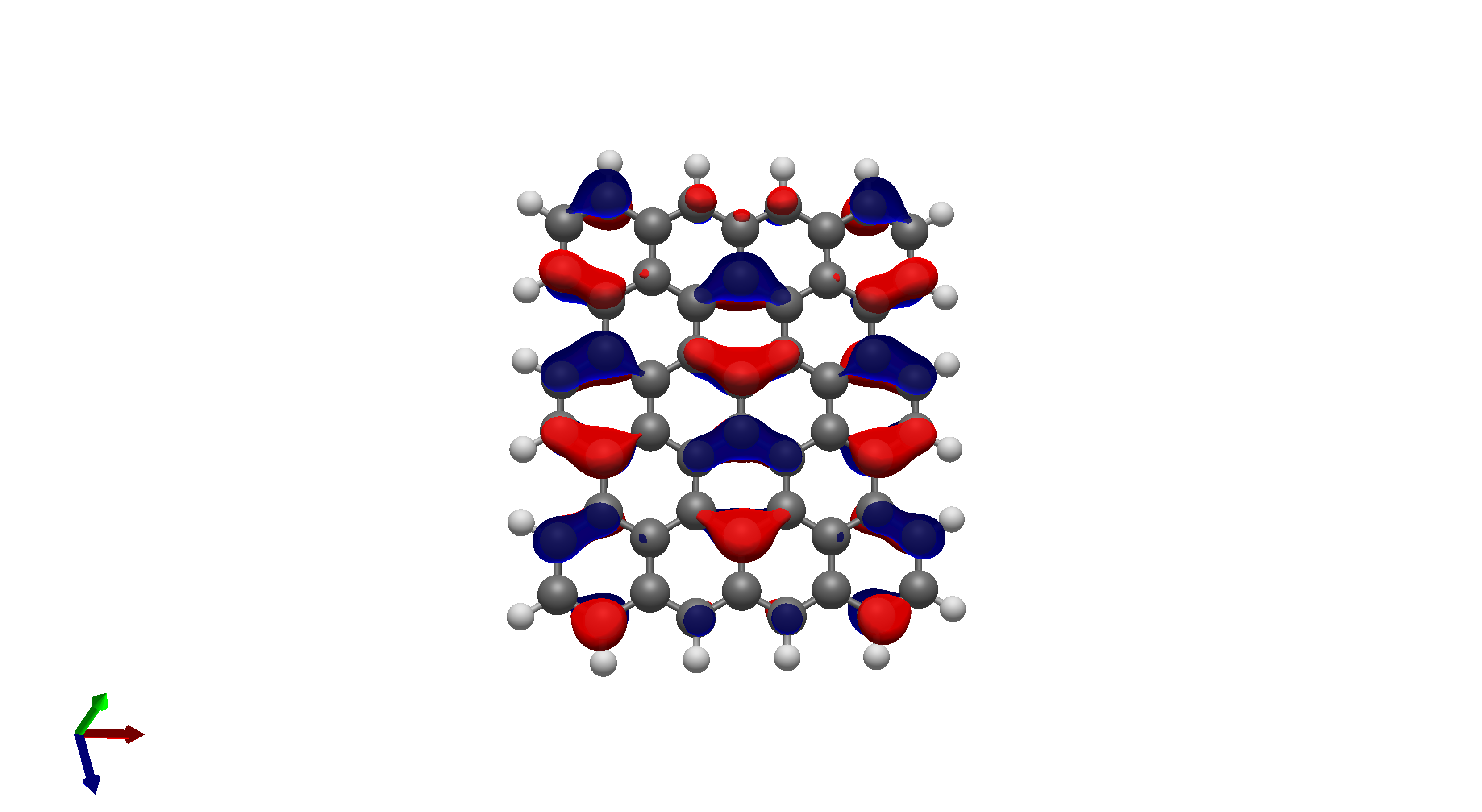
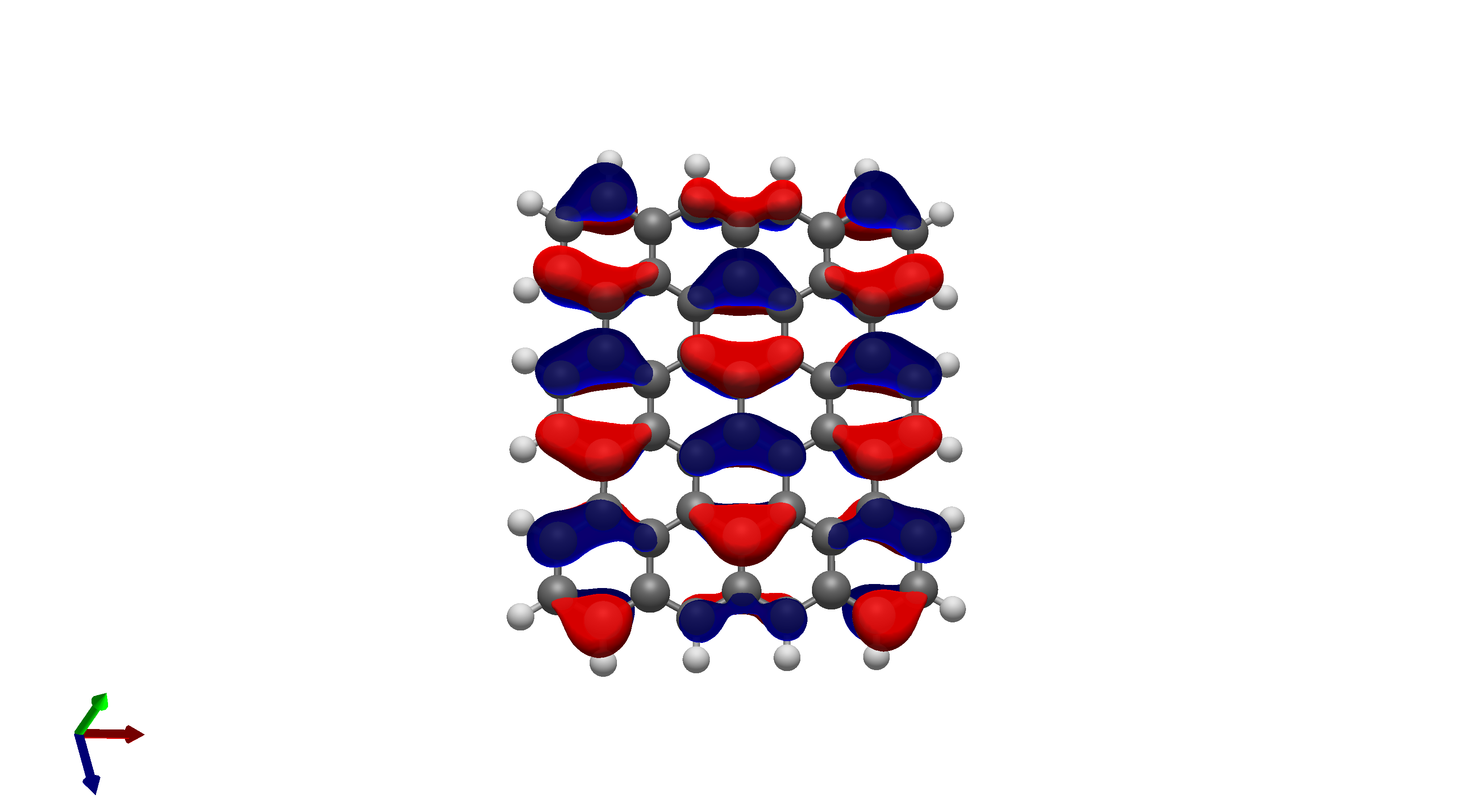
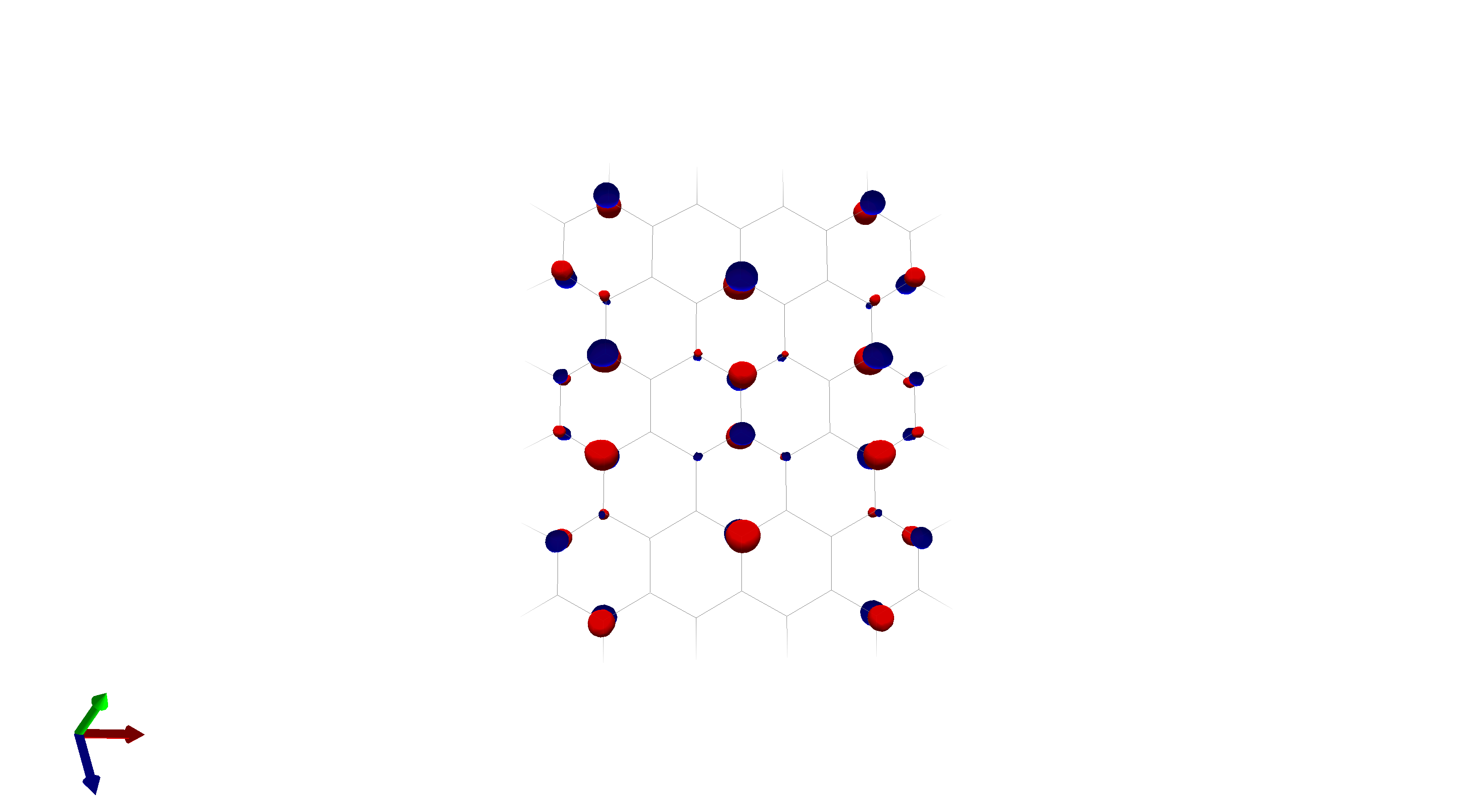
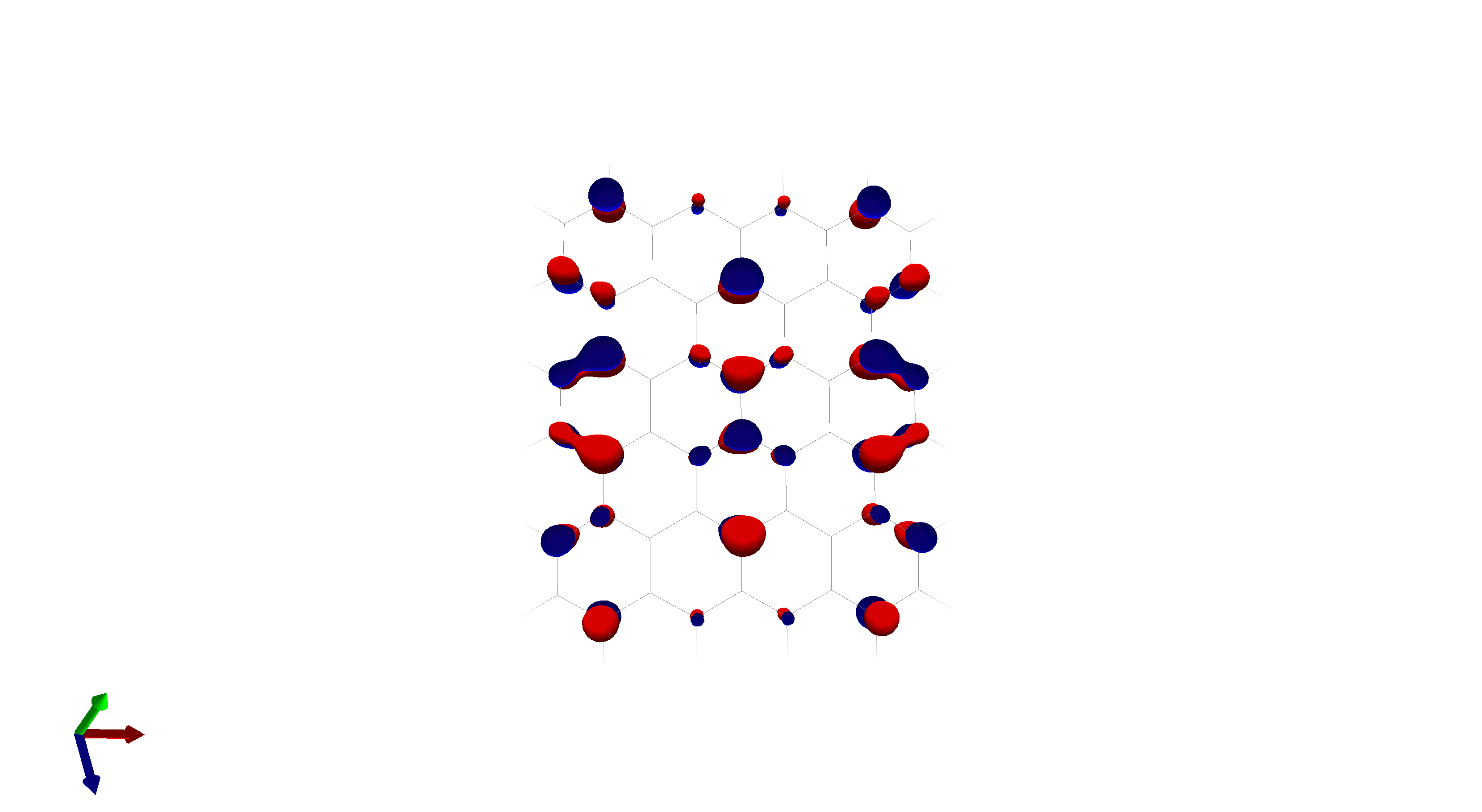
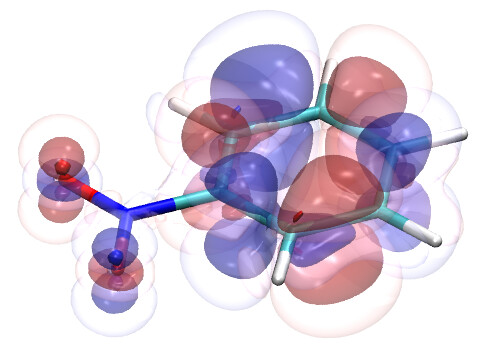
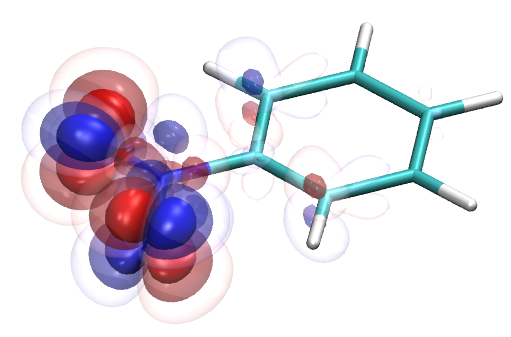
To this end, I’ve spent a (very) short amount of time getting pictures of values ranging from 0.08 to 0.01 for bis(fluorosulfonyl)imide (FSI), and 0.04 to 0.01 for a chunk of graphene (anything higher than 0.04 had orbitals smaller than the atoms themselves). The pictures from these are below, and I think there’s merit to a wide range of values.
In what is likely a controversial take, I’d really prefer that the values in Avogadro are limited to a discrete set of possible isosurface values, or at least there is a set of recommended values that a user could pick from. The reason for this is that in publications there is not necessarily a set value for isosurfaces, which can lead to significant misinterpretation of molecular orbitals in theoretical studies.
Of course, I am 99% sure that most of this will be a moot point once the volumetric rendering hits the shelves, but for now it’s something to be considered.
Perhaps we set a “spinner” that defaults to say 0.025 and has increments of 0.005. In general in Qt you can manually edit the number, but would be useful.
As far as the MO window, I directly ported from Avogadro 1.2, so I didn’t notice the default value was different. Happy to tweak it.
Might be useful to check defaults in GaussView and IQmol, for comparison.
(Where did the 0.0266 value come from? Seems very specific…)
For (small) molecules I would prefer to have a higher value (I have used 0.07 before). But I think in general 0.3 strikes a good balance for being applicable for multiple systems.
I have also seen people use 0.05 (or as they put it, containing 95% of the electron density), which would also make sense to me. It is always an arbitrary choice, but the 95% CI might inspire this.
When I first read the title of the thread, I thought it would be focused on the color (value) of the isosurfaces. In my opinion, there are better choices for the standard isosurface coloring than red and blue. Like blue and orange (#4286f4, #e29922), purple and yellow (#998ec3, #f1a340)
I’d be hesitant to use 0.05, since in my experience with trying 0.05 in the graphene chunk example you could hardly even see 0.05 with a ball and stick model. I have attached the wireframe version of the 0.05 isovalue, followed by 0.04, then another 0.04 but with the ball and stick model. It’s not the greatest comparison, but I think it illustrates the idea pretty well.
If I had to pick a default, I’d say 0.03 is good, and to the point of @ghutchis, I’d be thrilled to see an increment of 0.005 instead of the current allowed increments of up to 0.0001.
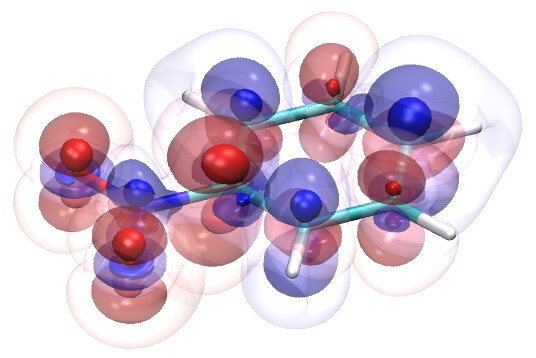
This came from a conversation I had with Dr. Jan-Michael Mewes, who suggested either 0.08 for small molecules or 0.0266 for large molecules based off of his own testing. He also shared this project that someone in his group worked on many many years ago for visualizing molecular orbitals, which I think is a good way to represent the diffuseness of orbitals in a pseudo-volumetric fashion. The TL:DR of it is that they picked a set of three isovalues, 0.0128, 0.0032, 0.0008, and then rendered the subsequent molecular orbitals with varying levels of opacity to simulate the output you’d expect out of a true volumetric render. Here’s some graphics from that blog post:
I think that this is a good approach, although I think it would be rendered largely defunct by the volumetric rendering that will hopefully be implemented soon.
Yes, I’ll be merging for Linux and Windows soon. Mac will sadly have to wait since Apple’s support for OpenGL is mixed (i.e., we have to do more work on our end to clean up old rendering code on Mac).