Quite often I find myself in a position, in which I want to rotate a molecule so that it stays within the same plane.
For example, I could have a geometry for \ce{R-M \dots A-B-C} and I want to rotate \ce{A-B-C} so that I have \ce{R-M \dots C-B-A}. It’s somewhat hard to achieve that with the current rotation tool, as only 10% of my effort goes into rotation in the desired direction, and 90% of the effort is lost trying to handle the molecule wiggling out of the plane. Maybe I’m not using the rotation tool properly, but I feel there’s a case for restricted rotations (say rotate within xy only, or yz, or xz).
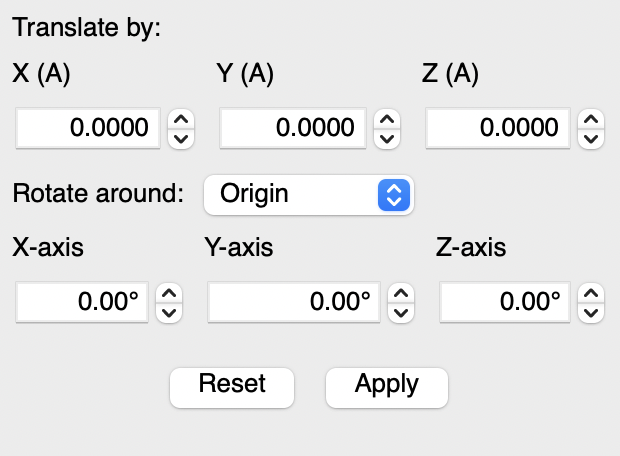
Yeah, I need to add back the manual rotation dialog from Avo1. You could easily translate in x, y, z a specific amount or rotate around the x, y, z axes.
It’s not perfect - sometimes you want to define a set of atoms as a plane and rotate around that arbitrary axis (e.g., rotate a Cp ligand) but it’s a known issue. I can probably find the time to get that in the next few days.
It already is possible to mark a molecule (or a group of it) and to initiate rotation (e.g., by arrow keys, shift and arrow keys), zoom-in/out (shift up/down), pan (e.g., Ctrl and arrow keys). Since I understand @seal’s search for a function is more like to mirror it vertically/horizontally, would it be possible to select the one molecule and subsequently perform these reflections by a short cut (like e.g., in ChemDraw sketcher)?
It already is very handy that Avogadro equally offers the home row as vim-like interface to the effect hjkl alone or with shift/Ctrl provide a gradual adjustment. Thus, Ctrl + l / Ctrl + r for a vertical mirror to the left or right, and Ctrl + u/ Ctrl + d for the horizontal mirror up/down could be short cuts currently (apparently) not yet used by other options in Avogadro.
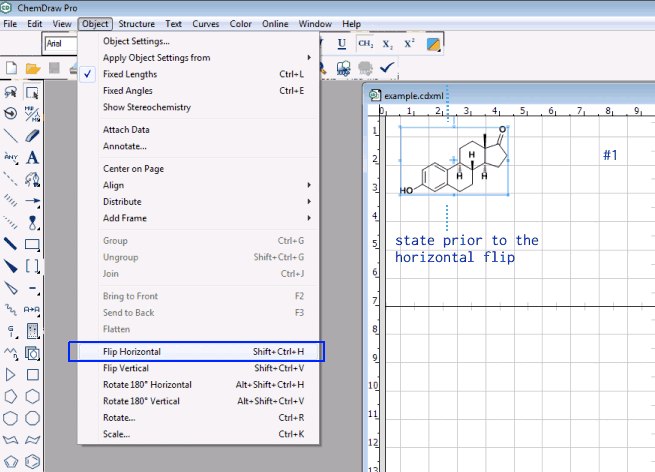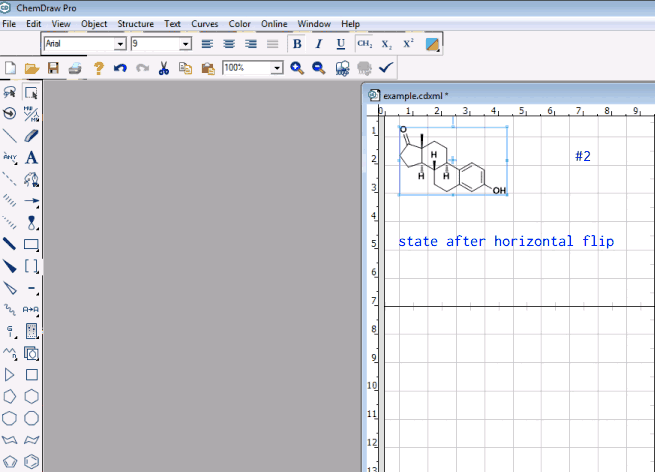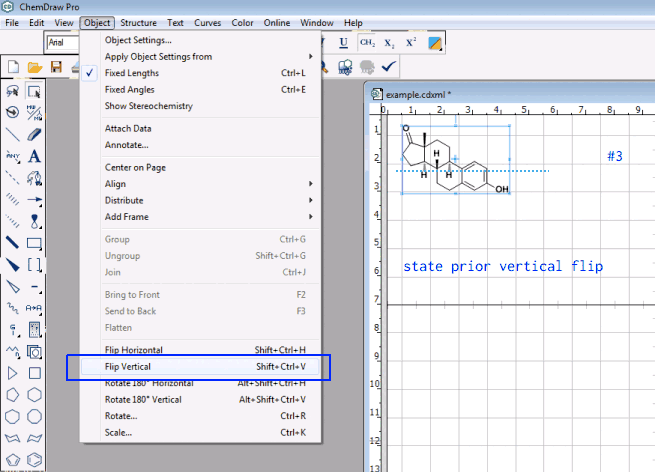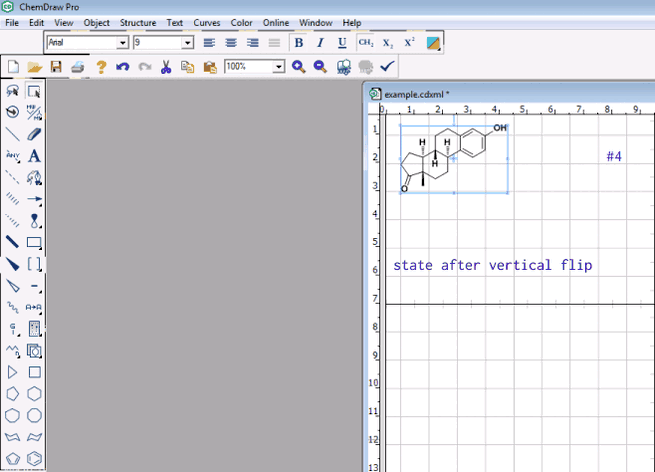
My suggestion was about using a mirror to reflect the molecule on a plane passing either the center between the extrema top/bottom, or most left/most right as provided e.g., by ChemDraw:
though the program equally offers rotation by 180 degrees clockwise/anticlockwise, or by any integer angle of the object selected (Ctrl + R), too. @seal’s comment by September 2nd, the thread likely is more about the later.
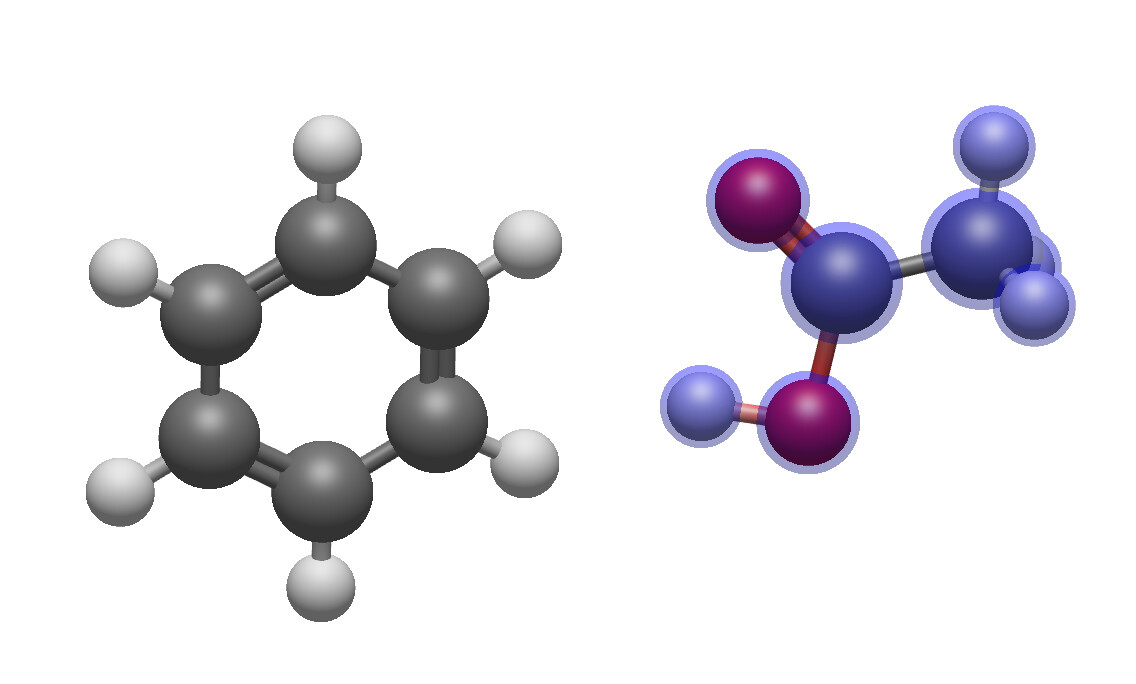
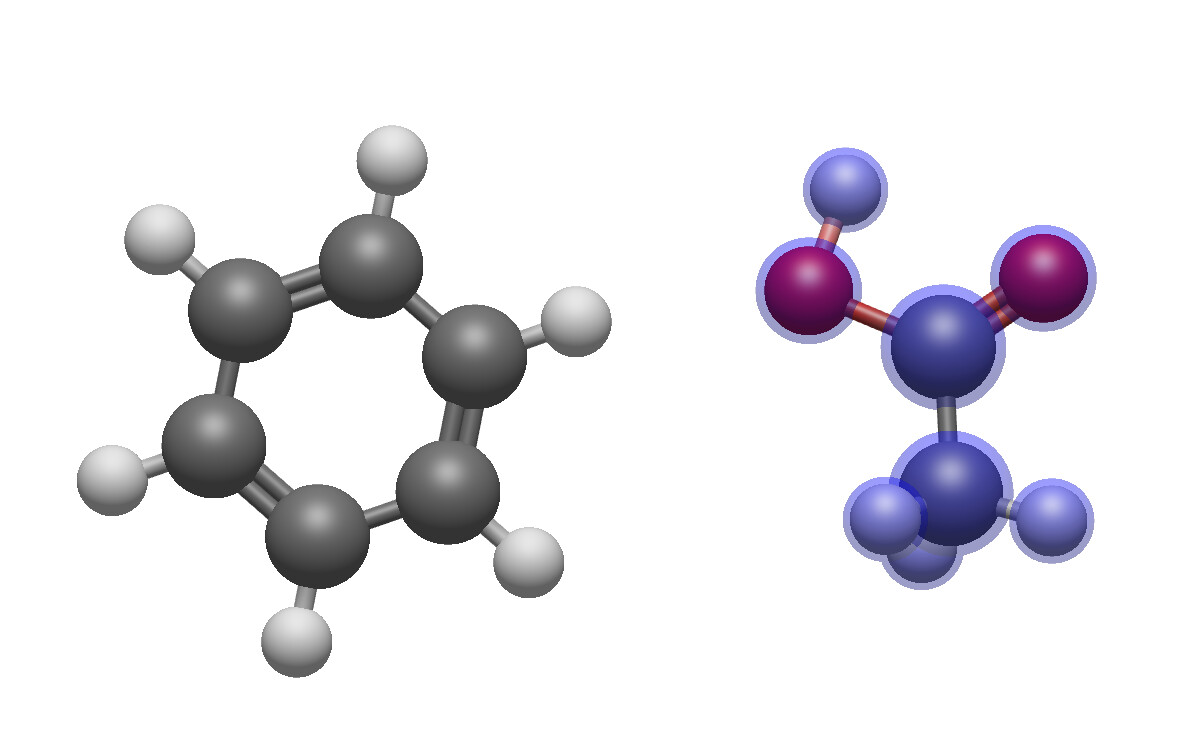
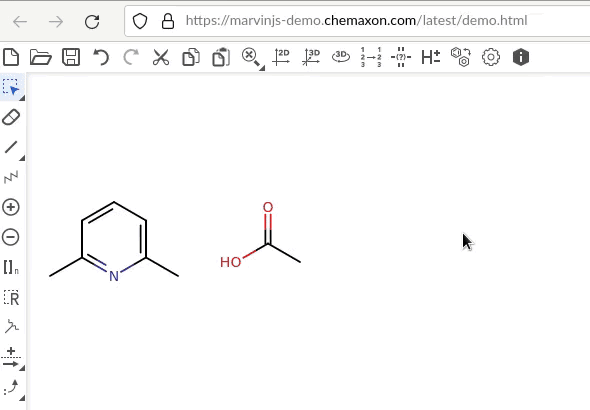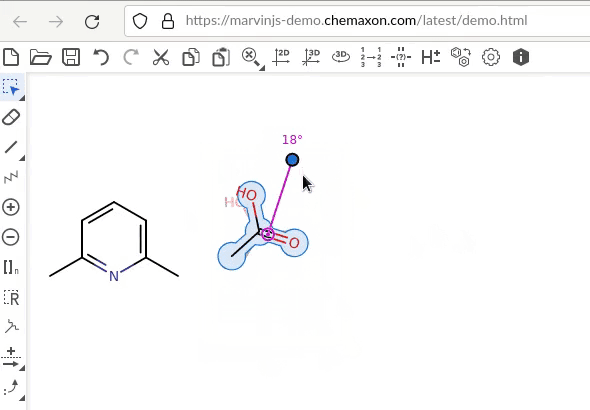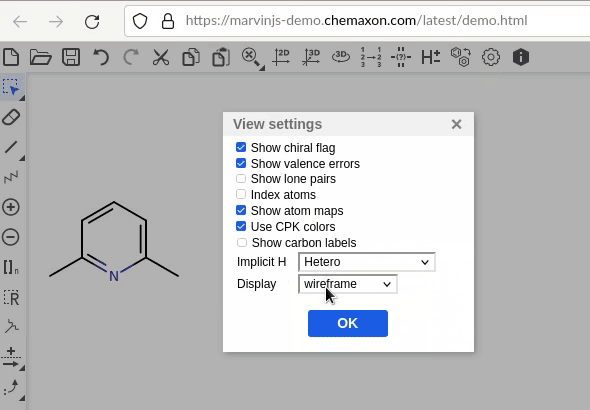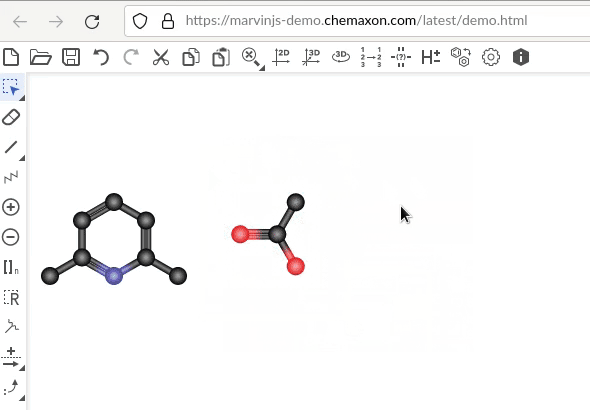
In both examples, the acetic acid was rotated. The difference is that in the first pair – assuming the C of the carbonyl group as reference for the acetic acid and the “north eastern” carbon atom of benzene for benzene – there equally is a shift/translation downward. In the second pair, the two atoms keep sharing their level.
So I think what you look for is a rotation of the selected molecule where the hinge where around which the rotation takes place does not change its position. There is something similar in ChemDraw with either fix 180 degree clockwise/counterclockwise rotation or any rotation of a dialed integer. In the skecther Marvin JS, it looks it’s action like this
and you possibly want to mark the molecule, and either enter a short cut like Ctrl + R to enter the angle manually, or a short cut likt Ctrl + Left (or Ctrl + Right) as in pdf viewers to rotate the selected molecule by a fix increment. If for the later, what would be the increment of preference be? ChemDraw’s default of 180 degrees is a bit large, at this stage of brain storming, one may reflect e.g., about steps of 90 or perhaps 45 degrees instead.
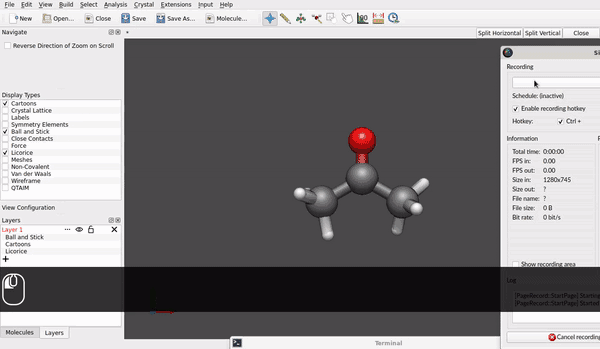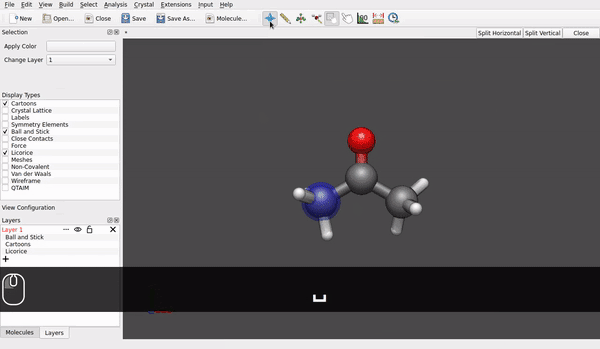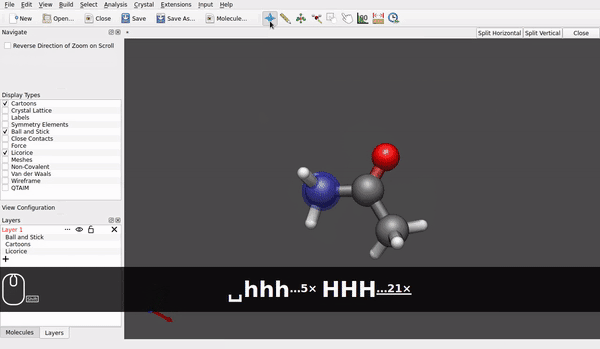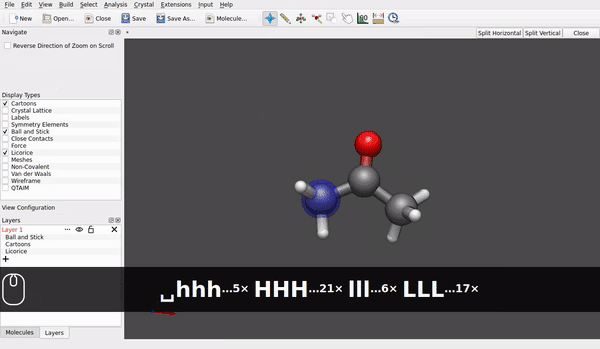
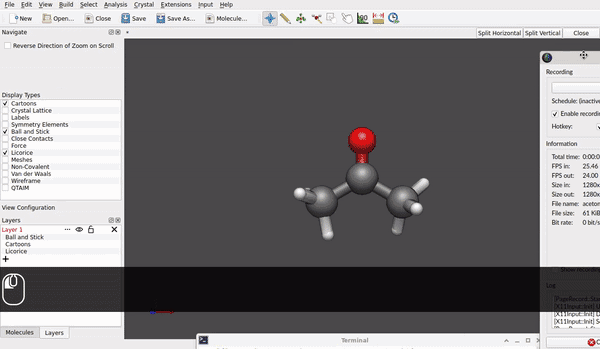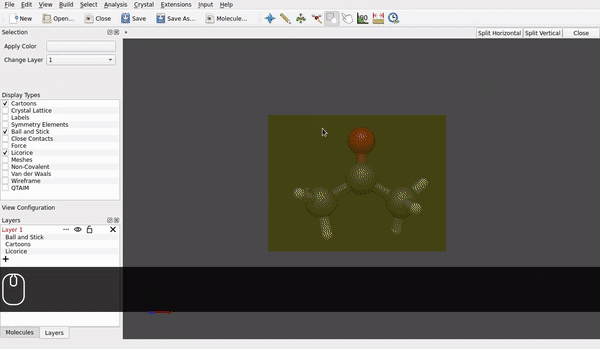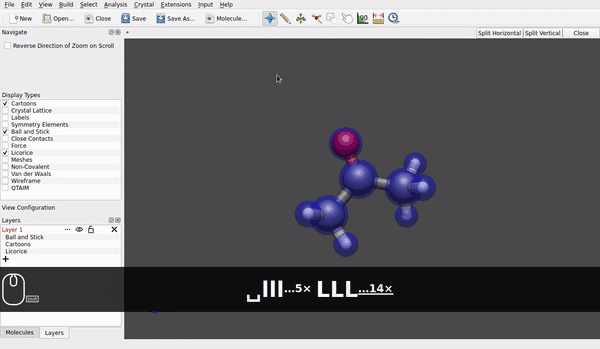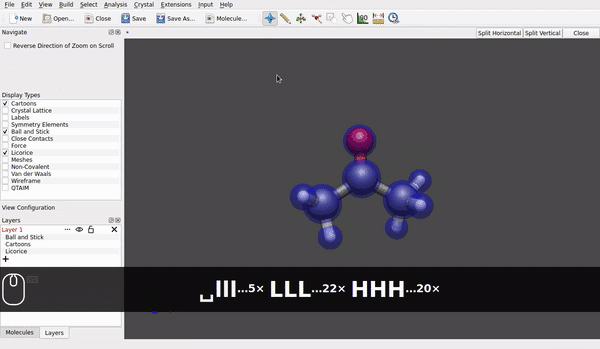
@seal Speaking for the AppImage for Linux downloaded today* and used in Linux Debian 12/bookworm (at present, it is branch testing) it looks like rotation of a molecule around e.g., a carbon of a methyl group already is possible (small h/ell and capital H/L with the shift key):
Maybe the hinge moves a little bit, but it looks different to rotating the acetone molecule as an ensemble: