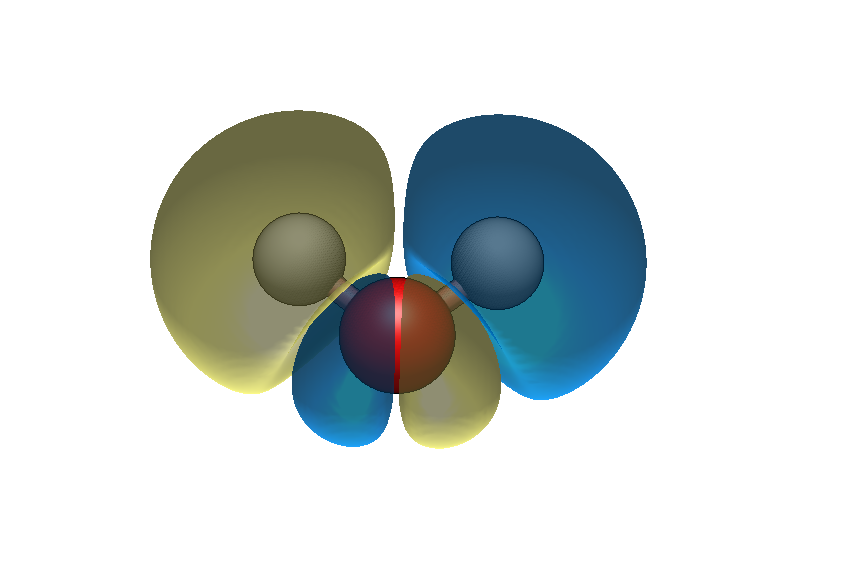I would like to use Avogadro to depict Molecular orbitals using the aux files calculated by OpenMOPAC2016 software.
The MO number seems to be invalid if I calculate MO surface in the “Analyze” → “create surface” menu. For example, water molecule has six valence MOs, 1 - 6. The MO 2,3,4,5,6 showed in the avogadro seems to be actually MO 1,2,3,4,5. Is this bug?
Environment Information
Avogadro version: 1.102.1
Operating system and version: windows 11
Thank you for your comments. Here I upload an MOPAC output file, H2O.txt. Please rename this to H2O.aux, and read it with avogadro. I can see the MO surface, but the MO number is incorrect.
MOPAC2016 eventually became OpenMOPAC with own public GitHub repository running under the permissive Apache 2.0 license scheme. Thus, beside official releases, the program equally is packaged by DebiChem (tracker page) and other platforms. Jonathan E. Moussa (the current lead developer) welcomes every interested contributor to the project.
Yep. I know - I helped Jimmy Stewart out a few times. At one point, he was getting NIH funding for MOPAC but needed a support letter saying that Avogadro would help providing a UI to visualize MOPAC results. I’m happy to see that it’s getting updates and maintenance now.
What I meant more was “I haven’t used MOPAC in a while, but I’ll definitely check it out.”
Certainly we should check to make sure that the MOPAC support still works with the most recent releases of OpenMOPAC.
And I’ll take a look at the MO numbering later tonight.
Thank you very much for your very fast work. I confirmed fixation of the software, continuous release, using the data of some simple molecules. I had been using Avogdro1 for studying the spectroscopic properties of some metal complexes, but (recently I retired from the research) I would like to use Avogadro2 for educational purpose.
You may also want to look at XTB / GFN2 for educational purposes. In general, I’ve found it to be more accurate, although it doesn’t directly give heats of formation.