Why are the atoms not correctly connected when an .xyz file generated by xTB is opened in Avogadro? How can this be corrected manually? Is there any documentation available on this issue?
In general XYZ files are supposed to have their bonding automatically interpreted, but perhaps you’re using an older version of Avogadro. Could you tell me what version you’re using, and what operating system you’re on?
Also, can you upload the XYZ file so I can take a look?
And it would help to know what you mean by “correctly connected.” No bond perception method will be perfect. XYZ files are simply a list of atoms and coordinates. It’s usually fairly easy to determine bond connections between atoms, but determining bond orders is sometimes very challenging.
thank you for your reply
By ‘correctly connected,’ I mean that the structure reconstructed in Avogadro bears little or no resemblance to the original molecule I drew in ChemDraw. For example, ring systems (such as three- or seven-membered cycles) may be formed when they should not be, and heteroatoms such as sulfur may appear unbound or improperly connected. This suggests that the automatic bond perception fails to reproduce the intended chemical structure.
Can you post the actual XYZ structure and/or the ChemDraw image you expect?
Otherwise it’s unclear what the issue is.
thank you for your reply
I am working on Windows 11, avogadro 1.2.0
Avogadro 1.2.0 is no longer supported, please try downloading the latest version (1.103) from the Avogadro 2 website and see if the issue persists.
same probleme with this version
Please do provide the XYZ file and/or images of the problem, it’s the only way we can point you in the right direction. You can just drag and drop the files into a post reply and they should upload.
They’re trying - unfortunately, the forum is marking as spam and I have to head to lecture. I’ll sort it out in an hour or so.
In the meantime, try going to Build > Bond > Bond Atoms and see if that puts bonds in the right places. If you’d like to try and have Avogadro perceive double and/or triple bonds, you can use Build > Bond > Perceive Bond Orders. If that doesn’t work, make sure your structure has the right total charge and spin, then try again.
@Nai tried to post, but as a new user posting attached files, it was flagged by the forum. I have no idea why I cannot undelete them - I’ll post a bug report with Discourse.
Here’s the ChemDraw:
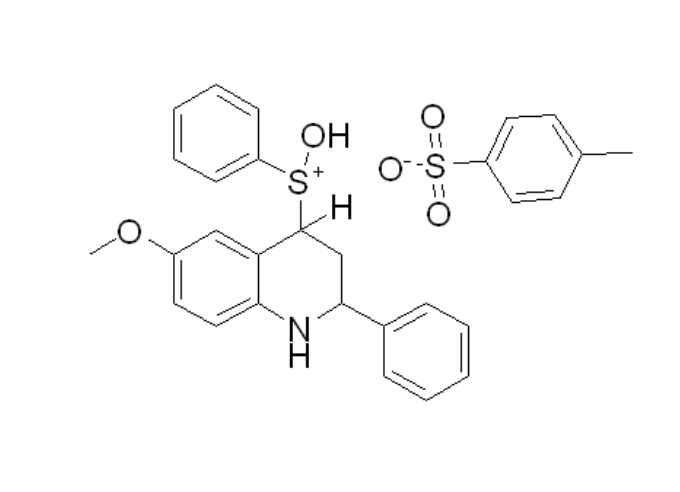
Here’s the file:
xtbopt.xyz (1.8 KB)
I don’t know how you tried to run XTB, but I immediately see the problem with the XYZ file. There are no hydrogen atoms. I’m actually impressed it ran at all - of course all those S, N, C, O.. they’re going to form bonds with whatever is near them. It’s not a problem with the bonding perception - just that the calculation ran without any hydrogens.
In ChemDraw, you can Copy As ⇒ SMILES and then in Avogadro Build ⇒ Insert ⇒ SMILES and paste in the text. This should give you a fairly decent 3D geometry to start.
Here’s a plausible XTB output:
xtbopt.sdf (6.2 KB)
I’d be curious to know how you got the structure into XTB though.
Thanks for your suggestion regarding ChemDraw SMILES and Avogadro, that’s helpful. My goal was actually to visualize the molecule after optimization rather than rebuild it from SMILES. However, I think I may be missing something. You mentioned the file xtbopt.xyz containing 37 atoms, but I don’t seem to have such a file. In my case, all the structures include hydrogen atoms, since we used SMILES in RDKit to add hydrogens before running xTB. Could you share please how did you do to run xTB to get this output?
I was looking at the file you uploaded to the forum. I’d suggest looking at both your input and output XYZ files in Avogadro.
As I said, I copied the SMILES from ChemDraw, used Build ⇒ Insert ⇒ SMILES in Avogadro which creates an initial 3D geometry including all implied hydrogen atoms. I exported this to SDF and ran XTB.
I’d double-check to make sure that whatever you’re doing in RDKit including saving the 3D geometry is including hydrogen atoms.
Thanks for your helpful reply. I believe the issue is more related to the file format than to the hydrogen atoms. I am currently using .xyz files instead of .sdf files.