Hello Everyone,
I am very new or not from quantum chemistry background, however i need some kinetic and thermodynamic data (Activation energy, reaction rates…) for my degradation analysis of fuel cell catalysts.
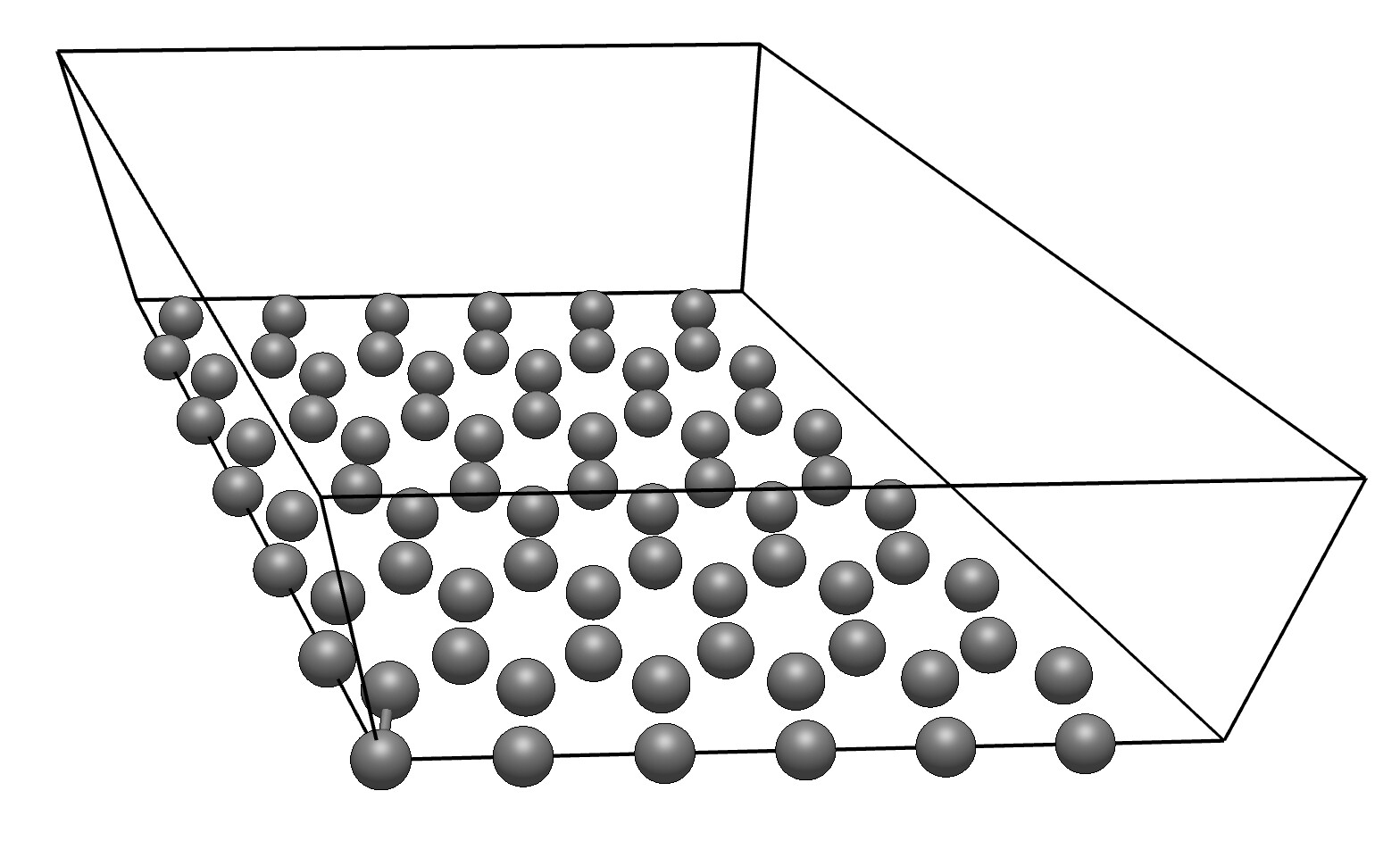
I am trying to find the reaction paths, transition states for the oxidation of solid carbon by water. I have found one literature where they have mentioned the structure of the slab however it was modeled in VASP. The slab model is as follows
‘The 6 x 6 optimized graphene supercell was in a hexagonal lattice with a =b =14.76 Angstrom (the unit cell vectors a and b in the surface plane). Repeated slabs were separated by a vacuum region of 21 Ansgtrom which is large enough to avoid interplanar interactions. A 2 x2 x 1 Monkhorst-Pack k-point
mesh was used for the Brillouin zone.’
Ref: Redirecting
How can i do such 2D graphite slab in Avogadro. I kindly ask your support and i thank you in advance.
There are a few ways to create a 2D sheet of graphite. Here’s how I’d do it, though:
-
Use File ⇒ Import ⇒Crystal and search for “graphite” or “C” to import the graphite crystal structure.
-
Generate the 6x6 supercell.
- In Avogadro 2, this is under Crystal ⇒ Build Supercell… (a = 6 and b = 6)
- In Avogadro 1.2, this is under Build ⇒ Super Cell Builder … (a = 6 and b = 6)
In Avogadro 1.98.1, this looks something like this. (I haven’t reset the bonds)
-
Increase the “c” axis distance to increase the vacuum region. In Avogadro 1.98, that’s Crystal ⇒ Edit Unit Cell … and change the “c” parameter to 21 Å (or whatever you want)
-
Export as POSCAR for VASP or set up the calculations as you desire.
Hope that helps!
Thank you very much. I also was researching in the background and i came up with the same solution. However, the crystal from the import ribbon, is it already optimized? Do i need to optimize the geometry again after building the supercell?
That depends on your goal. The geometry will be based on the experimental crystal structure.
It will probably not be the optimized geometry for whatever DFT method you pick, but sometimes you want to compare the computer to experimental geometry.