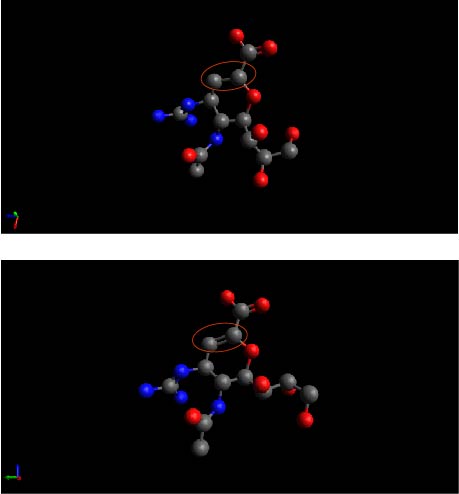
I have two structures which are complexes of a chemical (called ZMR) and a protein. I extracted the two ZMR from the two complex structures and loaded them into avogadro, respectively. The image of these two ZMR is attached at the following link:
Please note that the bonds in the red circle. In one ZMR structure it is a single bond (up), which in the other structure it is a double bonds (down). I wonder whether they are really different bond types or the difference is artificial associated with avodagro display. If they are really different, the how does avogadro discrete single or double bonds?
You haven’t mentioned how, specifically, you “extracted” the ZMR - presumably from a PDB file?
In general, PDB files do not record bond orders, so Avogadro (and the underlying Open Babel library) have to guess what bonds are single, double, triple, etc.
As you note in this case, the difference is visual.
If you have a link to the complexes in PDB, that would be great - we’re working on an improved bond perception code using LigandExpo, so I’d be curious to look at this test case.
Thank you for your response. The ZMR is extracted from pdb files. Becasue the complex structures have not been published, I am afraid it is inconvenient to link it to you now.
I have change the single bond in the red circle in the up molecule into double bond and now the bond orders of the molecules are the same. I now trying to add hydrogens to them with the "build>add . Strangely, only 10 H are added to the up molecule while 20 H are added to the down molecule. I think the two molecules are the same, then the numbers and positions of H added should also be the same. I wonder why are they so different.
I wish to do molecular dynamics simulation on these two complexes. But with such a large difference with H, I double the results of the simulation can be believable. Could you tell me what are the principles of avodrago to add H?
I am glad to see this post because I cannot optimize structures with intermediate bonds. For example, part of the molecule is H-C -(C-O-)2 and the two C-C distances are equal (1.395 A in crystal structure). Avogadro puts one as single and one as double. Then I enter the intermediate bond lengths and save. I optimize using ORCA and the final structure again has one single and one double bond, which is incorrect. This problem occurs many times.
I mean, the assignment of bond orders for visualization is never going to be 100% - particularly in your case. You can always “draw over” a bond in Avogadro to change the assigned bond order if you don’t like the appearance.
I suspect if you measure the bonds in Avogadro, you’ll see that they’re equal in geometry - it’s just that the bond order assignment code needs to pick one double and one single to fit the standard bond valence.
I don’t understand what you mean - if you import a file, the positions of the atoms will not change, no matter what the bond type displayed. Can you post the file?
Can I e-mail it to you? It is 58 MB. Orca has optimized C-C and C=C instead of C C intermediate. Strangely, Avogadro puts a single bond on the much shorter distance and a double bond on the much longer distance.
You can definitely e-mail me, use PasteBin, or other link. Ideally if you’re going to e-mail either put it up on Dropbox / Box, etc. to download, or use gzip/bzip to compress before sending e-mail.