We’ve been releasing beta versions of Avogadro2, which should be finished sometime this fall. The latest is 1.94 (https://two.avogadro.cc/)
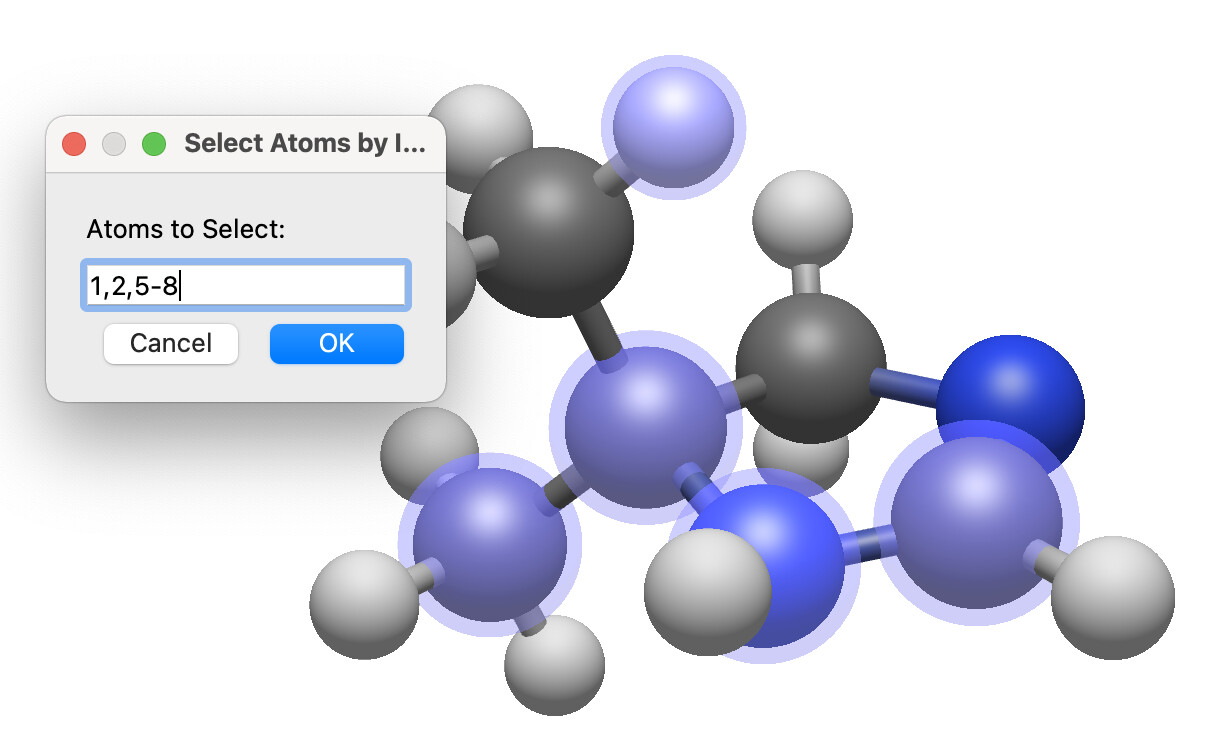
As part of that, I just submitted a patch for selecting a comma-separated list or range (or both), e.g. “1, 2, 5, 6” or “1, 5-10”:
While Avogadro2 isn’t feature-complete, it’s easy for us to release builds from patches for people to try.