Dear Michael,
I assume you equate “atom number” with atoms of either carbon, or nitrogen. Perhaps not a solution for Avogadro, but if your aim is the rapid identification of atoms of a particular type, an intermediate alternative may be to process your structure model by Jmol. Among the input formats supported are .xyz, .pdb, .cif, .mmcif; if not available for you by default, .xyz and .pdb are formats OpenBabel (here is the current documentation) may write for you from many other formats.
Once the structure is read by Jmol, open the program’s own script console. Here, 1) select the objects of interest, and 2) assign the colour. For example
select oxygen;
color green;
will substitute the default red about oxygen atoms by green. The commands may be extended, e.g., to select multiple atom types at once
select oxygen, nitrogen;
or to select larger (possibly recurrent) pattern like
select leu; # selects all leucins if assigned in the .pdb
select protein; # selects assigned proteins in a .mmcif
In terms of colours, you may use instead an RGB definition, too:
color [123, 255, 13];
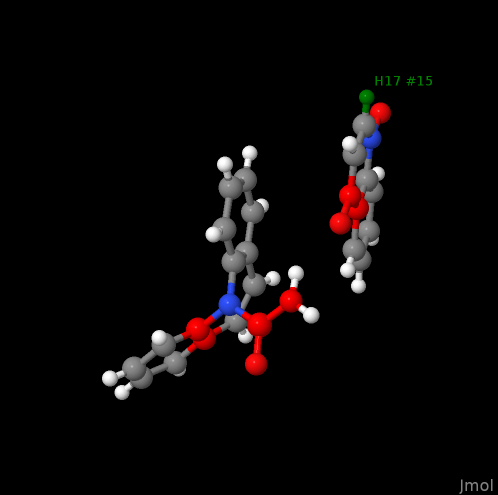
As an illustration of the above, with the .mmcif of 1NE6, for example,
select arg; # selection of all arginines
color [255, 255, 255];
write "arginines.png";
yields the following:
while the calls of
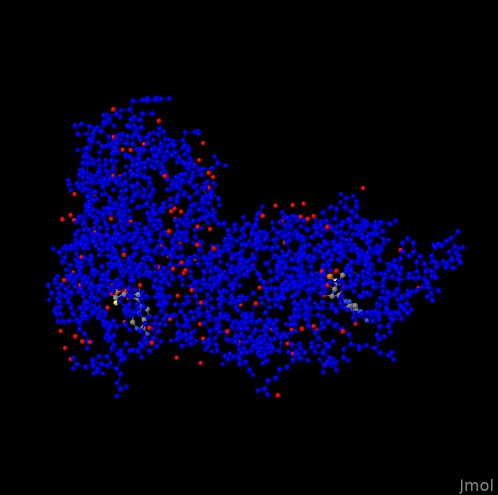
select protein;
color blue;
write "protein.png";
alter the visualization to
which highlights water and guest molecules.