I noticed that while CJSON files are exported with coordinates to 14–16 decimal places, XYZ files only get 5.
In comparison ORCA and xtb both write 14 to file. Can Avogadro match that?
I noticed that while CJSON files are exported with coordinates to 14–16 decimal places, XYZ files only get 5.
In comparison ORCA and xtb both write 14 to file. Can Avogadro match that?
Why do you need so many decimal places? Certainly the position of the atoms is not accurate to 1.0 \times 10^{-14} Å. Maybe, just maybe the convergence with a good quantum program is accurate to ~1.0 \times 10^{-5}Å.
CJSON simply uses the default for JSON export. I don’t believe any current JSON library lets you set the precision or I’d do that to save space (e.g., Is there a way to control the precision of serialized floating point numbers? · Issue #677 · nlohmann/json · GitHub)
Can you find me an example in which more than 0.00001 Å accuracy is required?
Obviously that ridiculous level of precision is meaningless in a final result. But that’s not the point.
It’s primarily about avoiding rounding errors. Rounding shouldn’t be done until the end. Avogadro can’t and shouldn’t assume that exporting to this or that format is the final step in the user’s workflow and therefore is the appropriate time to do rounding.
If anything, I think producing an XYZ is most likely to be the first step of a workflow not the last.
Even as a single example, if I start an xtb optimization with a crude structure with an XYZ file with either 5 or 14 decimal places, the resulting optimized geometry differs in many of the coordinates in the fifth decimal place. In several cases the difference is greater than 0.00003 A, so the original rounding error is already compounding. This has consequences for reproducing results.
And multi-step calculations will only see further compounding. Sure, the user can use tighter convergence criteria, but the larger the differences in input structures are, the more likely it’ll end up changing something as important as which minimum is reached, right?
Obviously super accurate geometries are not all that important, that’s why it’s broadly fine to use cheaper methods for optimizations and save the expensive methods for single points. But why risk it? Why withhold data that Avogadro has readily available?
It’s also one of principle: Avogadro and other programs shouldn’t throw information away on behalf of the user.
For example, data should be preserved across I/O cycles (to the extent that is viable). If a user loads an XYZ with 10 digit precision, changes a single atom’s position or adds a methyl group or whatever, then saves a new XYZ, why should the user lose information across that process?
Especially because it’s not even “loss” of information, it’s deliberate omission on Avogadro’s part.
What if a user runs a calculation to find a transition state and the program they’ve used gives them it in format (a) with high precision, but then they have to convert it to format (b) for use with some other program that doesn’t accept (a). Isn’t a bad thing that the user would then get a different result depending on whether they used Avogadro to do the conversion or some other tool?
Besides, XYZ files are tiny. ORCA and xtb both print the geometries with truncated precision in the coordinates within their output, I guess because with so many geometries it makes a difference to the size, but for the saved output geometry they use the higher precision. For a single geometry in XYZ format we’re talking about kilobytes and users would be free to reduce the file size that way themselves at a later point if it was so crucial to them.
If you think 14 is just truly unnecessary, then something like 8 or 10 would at least mean it can be reasonably assumed that results from subsequent calculations are accurate to 5 significant figures.
If you think a calculation will actually suffer from accuracy below 0.00001 Å, I’d like to see the specific example. In most cases, QM programs will end geometry optimization before converging to that point.
You seem to think that 10-digits actually has useful information. I agree that information should not be lost. But I also don’t think anything beyond ~5 decimal places in position is actually useful information vs. numerical noise.
Let me be clear. I’m a scientist and I’m absolutely willing to be convinced on this.
I just want data or an example in which there’s a difference.
I feel like you’re misunderstanding me, I’m not trying to argue that the 10+th digit is important information for us as chemists. I’m making the point that the result you get out of any function is different when the values of its parameters are different, and the effect compounds, so if you want two of the “same” calculation to give the “same” result in that they agree to e.g. 5 significant figures you need to start with numbers that agree to much greater precision than that.
I figure I’ll throw in my two cents here, since I often am concerned with precision in position for my research.
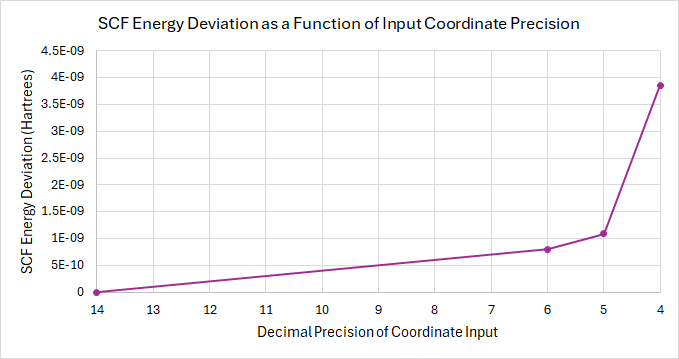
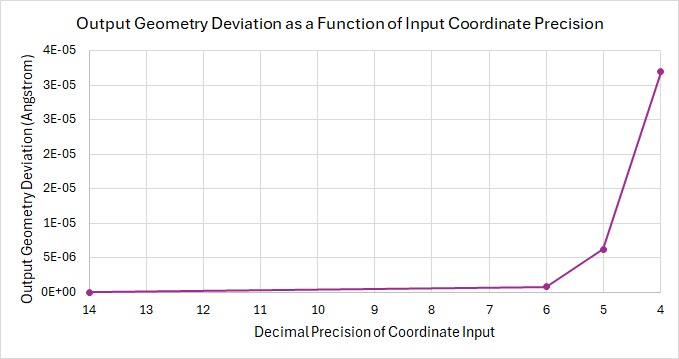
I ran an, albeit basic, test calculation that used coordinate inputs with 14, 6, 5, or 4 decimal points as the input.
My method for generating each input file was as follows:
Take the ORCA .xyz output from a previous calculation with 14 decimals, and copy exactly the coordinates into an input file. For 6 decimals, I opened that same .xyz file in Avogadro and used the ORCA input generator, which gave an output with 6 decimals. From there, I used that input as a template, and manually rounded off the coordinates to either 5 or 4 decimal places, making sure I didn’t do any double rounding.
The method used is shown below:
!Opt wB97M-V def2-QZVPP LargePrint DEFGRID3 VeryTightSCF VeryTightOpt
%pal
nprocs 20
end
I have included a tar file here that has the original .xyz file, the inputs for each calculation, the final .xyz for each calculation, and a CSV that summarizes three components of each calculation: final SCF energy, final coordinates, and total time.
The conclusions that I have drawn, and I am sure that the same conclusions will be drawn for the rest of us, is that the deviation in the final output energy and geometry is directly related to the precision of the input geometry. See the below graphs for a brief summary of these results.
Some important things to note are that I only ran 4 calculations, that were on an already-optimized geometry, and so these results only come from a stable point on the potential energy surface, and as such are not by any means extreme cases. I think that if the shape of the PES would drastically alter the results, giving the input precision much greater weight on the final geometry.
From these results, I suggest the same thing that @matterhorn103 does, that coordinates generated from Avogadro are printed with 10 decimals, which should greatly reduce the probability that the outcome of a calculation is significantly altered by the coordinate input precision.
You’re expecting agreement of the order of 1.0e-9 Hartree. Let’s turn that into kcal/mol = 627.509 kcal/mol per Hartree. So that’s an accuracy of … ~6.275090e-7 kcal/mol?
You have way, way bigger errors in your calculation, even at VeryTightSCF, etc.
Try switching processors. Or you probably use the ORCA binaries… Try compiling QM code from source and switching different compilers.
Floating point errors < 1.0e-9 are close to numerical noise. Most QM programs avoid some integrals at that threshold. Most programs will tell you that two floating point numbers are identical at that precision.
And then you’re indicating that there are deviations in the output geometry of ~5e-6 Å because of a roundoff of five decimals in the input coordinates (i.e., the deviation you claim indicates exactly the number of significant figures in the XYZ file).
As I said, I’m willing to take a patch or see data that there’s really a meaningful difference because of the decimal precision in the XYZ files.
But I think your data is basically saying what I am. That there’s insignificant differences at that precision.
I feel like the thing to take away from this is not that the deviations are insignificant, but rather that they exist at all. If you have an unstable location on the PES then these changes are amplified, and furthermore, the associated cost with changing XYZ files from 5 decimals to 10 is extremely low.
See the files I’ve attached. A 8500 atom XYZ file with 5 decimals is 314,612 bytes (coordinatetest.xyz), versus the same file but with 10 decimals is 476,006 bytes (coordinatetest2.xyz). There is only a ~1.5x increase in file size for doubling the decimal precision. Furthermore, if instead of using a bunch of spaces for the XYZ file writing it were instead using tab characters, the file sizes would be cut down even further to 239,231 bytes for 5 decimals (coordinatetest3.xyz) and 366,731 bytes for 10 decimals (coordinatetest4.xyz).
coordinatetest.xyz (307.2 KB)
coordinatetest2.xyz (464.8 KB)
coordinatetest3.xyz (233.6 KB)
coordinatetest4.xyz (358.1 KB)
I think my question is: Even if it might not be chemically significant changes (which I still think for very subtle structures it could be extremely significant), what is the downside?
Ok, I’ve run a bunch of short calculations on a variety of test structures to try and persuade you that Avogadro’s behaviour might make a difference.
What I have done is draw unoptimized molecules by hand in Avogadro, as you might for preparing a file for a calculation. I save a single geometry file for each test molecule containing coordinates to 14+ decimal places, and load it in Python. I then round all the coordinates to the specified precision, which I’ve set at 5 for now since that’s the status quo and the level we’re discussing.
To then simulate what difference variation at that level of precision can make, I generated 100 sets of coordinates where each set of each coordinate had a 50% chance of being increased by 0.00001 angstrom vs the original.
The average rounding error introduced by rounding to 5 decimal places is not generally as high as 1e-5 of course, but it’s of the same order of magnitude and this exercise serves to demonstrate the impact it can have.
I then ran an opt+freq with xtb on all 100 slightly different structures, per test molecule, analysed the results with polars, and compared the variation in various values in the output.
For something like gycerol the variation is not all that huge:
glycerol
┌────────────┬───────────┬───────────┐
│ variable ┆ std ┆ range │
╞════════════╪═══════════╪═══════════╡
│ energy ┆ 1.7758e-9 ┆ 8.1450e-9 │
│ coord_mean ┆ 0.000003 ┆ 0.000012 │
│ coord_max ┆ 0.000006 ┆ 0.000023 │
│ freq_mean ┆ 0.001882 ┆ 0.008211 │
│ freq_max ┆ 0.00884 ┆ 0.0401 │
└────────────┴───────────┴───────────┘
To explain what these numbers actually mean: I collated the values for the final energy, all of the individual x, y, and z coordinates, and all of the individual frequencies. I then looked at the std error and the range of the results for each individual value. Because there are tens or hundreds of different coordinates, and tens of frequencies, I then found the average std error and average range, and the maximum for each, across all coordinates, and across all frequencies.
So what the table above is saying is that across 100 runs of a glycerol opt, the std error in each coordinate value was on average 3e-6, in the worst case 6e-6, while the largest variation in any of the coordinate values was a range of 2.3e-5. The frequencies also only vary by fractions of wavenumbers, and the energies always come out somewhere in a 8e-9 hartree range.
Benzene also unsurprisingly looks to basically withstand this degree of variation:
┌────────────┬───────────┬───────────┐
│ variable ┆ std ┆ range │
╞════════════╪═══════════╪═══════════╡
│ energy ┆ 1.4625e-9 ┆ 7.7370e-9 │
│ coord_mean ┆ 0.000002 ┆ 0.000011 │
│ coord_max ┆ 0.000004 ┆ 0.000019 │
│ freq_mean ┆ 0.029572 ┆ 0.21317 │
│ freq_max ┆ 0.3272 ┆ 2.3171 │
└────────────┴───────────┴───────────┘
Though the variation in the frequencies is significant, I guess because of the role of symmetry?
Unfortunately if we look at something larger the compounding of the errors worsens:
aspartic_acid
┌────────────┬──────────┬──────────┐
│ variable ┆ std ┆ range │
╞════════════╪══════════╪══════════╡
│ energy ┆ 0.000004 ┆ 0.000017 │
│ coord_mean ┆ 0.028839 ┆ 0.103577 │
│ coord_max ┆ 0.082202 ┆ 0.268131 │
│ freq_mean ┆ 0.784611 ┆ 3.892848 │
│ freq_max ┆ 2.97416 ┆ 17.4138 │
└────────────┴──────────┴──────────┘
Here we even have a variation in the single point energy that is approaching the sort of magnitude where it starts to make a difference (though it’s still below the level of being meaningful).
And now three examples that don’t optimize to a minimum but rather a saddle point of some degree (everything previously did optimize to a minimum):
acetic_acid
┌────────────┬───────────┬──────────┐
│ variable ┆ std ┆ range │
╞════════════╪═══════════╪══════════╡
│ energy ┆ 4.4624e-7 ┆ 0.000002 │
│ coord_mean ┆ 0.000457 ┆ 0.002148 │
│ coord_max ┆ 0.001112 ┆ 0.005523 │
│ freq_mean ┆ 0.190551 ┆ 0.868044 │
│ freq_max ┆ 0.759079 ┆ 3.426 │
└────────────┴───────────┴──────────┘
acetone
┌────────────┬───────────┬───────────┐
│ variable ┆ std ┆ range │
╞════════════╪═══════════╪═══════════╡
│ energy ┆ 2.2648e-8 ┆ 8.3869e-8 │
│ coord_mean ┆ 0.000027 ┆ 0.000103 │
│ coord_max ┆ 0.000077 ┆ 0.000294 │
│ freq_mean ┆ 0.138108 ┆ 0.966263 │
│ freq_max ┆ 1.521546 ┆ 10.8453 │
└────────────┴───────────┴───────────┘
dimethylurea_dimer
┌────────────┬───────────┬──────────┐
│ variable ┆ std ┆ range │
╞════════════╪═══════════╪══════════╡
│ energy ┆ 6.4671e-7 ┆ 0.000004 │
│ coord_mean ┆ 0.001024 ┆ 0.00979 │
│ coord_max ┆ 0.006106 ┆ 0.05883 │
│ freq_mean ┆ 0.096295 ┆ 0.812409 │
│ freq_max ┆ 1.594939 ┆ 11.8998 │
└────────────┴───────────┴──────────┘
What you can see is that sometimes the input geometry doesn’t make all that much difference to the geometry of the end result, or to its properties, and sometimes it really does.
My aspartic acid has 0.03 A variation in all the atomic positions in the output just due to randomized 1e-5 differences in the initial positions! And this is despite the fact that it reaches a minimum of the PES in all cases, so it is always in theory a “successful” optimization.
And the most extreme example:
ethylene_glycol_diacetate
┌────────────┬───────────┬──────────┐
│ variable ┆ std ┆ range │
╞════════════╪═══════════╪══════════╡
│ energy ┆ 0.001118 ┆ 0.002418 │
│ coord_mean ┆ 0.046022 ┆ 0.11918 │
│ coord_max ┆ 0.485238 ┆ 1.129222 │
│ freq_mean ┆ 4.01064 ┆ 9.839013 │
│ freq_max ┆ 83.425478 ┆ 182.9981 │
└────────────┴───────────┴──────────┘
Here we actually have an example where these tiny variations in input structure cause the qualitative result to vary, as the optimization sometimes lands at a minimum (69%) and sometimes only on a saddle point (31%)!
Obviously xtb is not very accurate. Obviously the use of tighter convergence criteria would have an effect, as would tighter grids, as would all the factors you mention like the compiler, the processor, etc. But none of them change the fact that what you put in directly affects what you get out, and sometimes significantly.
And these xtb structures would in most cases be used for a further optimization with DFT. The thing is, now the variation in different input geometries is not only on the order of 10^-5 per coordinate, it’s much higher. So the variation in the DFT results might well also be even greater, because the trajectory during the optimization is of course directly determined by the input geometry.
These are also all main group compounds in the set, nothing heavier than oxygen, no ions, no radicals, nothing hugely complicated. And there are a whole lot of things I didn’t test like NMR shifts and coupling constants, CD spectra, searches for conical intersections with TD-DFT, binding energies in supramolecular complexes, non-covalent interactions, where I don’t personally have any experience but I can imagine they could be quite sensitive to small changes in a geometry.
I want to make clear that I’m not trying to argue that the result of a calculation is made any better or any more valid by increasing the precision of the input geometry, and I’m also not trying to argue that a difference of 1e-5 angstrom is important in a final result. I just want to make the case that someone who prepares a molecule in Avogadro, then exports it to file, then runs a calculation on that file, shouldn’t get different results based on whether they exported to XYZ or CJSON or SDF or PDB or whatever. And opening an XYZ file in Avogadro then re-exporting shouldn’t result in a huge loss of precision, because we can’t rule out that happening as an intermediate step in someone’s workflow.
For comparison, the results with rounding to, then varying at, 14 or 10 decimal places are attached.
results_14.txt (5.6 KB)
results_10.txt (5.8 KB)
I also ran each calculation 20 times with the exact same geometry and the results are identical each time, so xtb is otherwise deterministic on my system at least.
But SDF only supports four decimals and PDB only supports three decimals.
e.g.,
https://www.wwpdb.org/documentation/file-format-content/format32/sect9.html
Of course. That’s related to the optimization convergence criteria.
@brockdyer03 brought up energy differences … on the order of VeryTightSCF in ORCA = 1.0e-9 Hartree. Which, of course.
You’re arguing that there are small differences in the xtb optimization:
https://xtb-docs.readthedocs.io/en/latest/optimization.html#optimization-levels
What you are both arguing, are that there are small numeric differences in calculations.
If we increased the floating point presentation to … say 8 decimal places, someone will come along and tell us that there are small numeric differences. Because there are small numeric differences in any sort of numeric calculation.
I’ll post my answer from Stack Exchange.
I have to say, as someone who doesn’t have really any real desire for either end of things, I just don’t see why not. I understand your point @ghutchis that the small to medium changes in the results from these calculations aren’t really significant. However, I do not understand your reluctance on just changing the output precision.
If you are concerned about precedent, that is understandable, but I can’t think of any other reason why this change shouldn’t be implemented. It certainly couldn’t be file size concerns, since increasing the precision of the coordinates is such a minor change, and any program that is going to take in the XYZ coordinate is already going to convert it to either a 16- or 32-bit float and just have a bunch of trailing zeroes. There also isn’t an existing standard for XYZ files, in fact most programs that output them will print out all 14 decimals that are associated with 16-bit floats.
I just am not sure why this change shouldn’t be implemented other than the fact that the result of such a change is potentially insignificant and thus isn’t worth the time. At any rate, the real reason for increasing the precision is to avoid losing data, as would happen if I loaded an ORCA XYZ output which then would be truncated.
Multiplied across the number of files times all the Avogadro users in existence.
You say it’s an insignificant change, but you haven’t run sampling across 3-million compounds or generated trajectories of thousands of frames of thousands of atoms.
And you might say “but then use a compressed format”. Yep. I want to do exactly that:
If it’s going to make y’all happy to increase the precision to say 8 decimals, please submit a pull request.
XYZ coordinate is already going to convert it to either a 16- or 32-bit float and just have a bunch of trailing zeroes
Do you know how floating point numbers are stored? Remember, they’re base-two.
So a 32-bit float has 24 bits of precision ~7.2 decimal accuracy. Yes, in principle a 64-bit double has ~14 decimal accuracy, but my point is that for atom positions, much of that is really not useful information for any practical calculation.
Why should we be insisting that ~2M downloads should continue saving a pile of useless bits for eternity? As I mentioned, SDF and PDB have even lower accuracy … because in reality, atoms aren’t at fixed positions – they’re dynamic.
If you really, really feel strongly. Fine – as I mentioned earlier – submit the pull request.
It wouldn’t really make me happy to add something to a program against the express wishes and views of the lead author of that program while that author vehemently disagrees with the suggested logic for doing so. If you, as an expert computational chemist, think I’m wrong, I don’t want it implemented. I don’t want to “win” by just banging on about it until you cave, I want Avogadro to have whatever the sensible correct behaviour is, because as you say, it affects many users.
I really don’t think I am. It’s hard to explain what I mean via a forum, but I’m doing my best to present well-constructed arguments. I feel a bit like you’re focussing on the aspect of what level of precision in an output geometry can be considered meaningful – where I don’t disagree with you! – and glossing over my point that different inputs lead to different outputs, and sometimes markedly different ones.
You originally asked
and I feel like I did this, so why does it not persuade you in any way? Yes, the GFN-xTB methods are not very accurate methods, and yes, they might not use default settings that lead to high accuracy, but they are real methods used by real chemists, so the ethylene glycol diacetate example I easily found shows that this is something we can expect to happen in the real world.
I’d have thought xtb was a good example of real world usage, of exactly the kind of thing that XYZ files produced by Avogadro will be put into; if the calculations had been run with DFT you might have said that it’s unrealistic to run DFT with structures taken straight from Avogadro without being pre-optimized.
Yes, semi-empirical methods are low accuracy, I was just endeavouring to give you statistically meaningful data without running hundreds of calculations on a cluster. Instead I’ll resort to a theoretical discussion.
For the sake of argument let’s say that actually people are only ever using the XYZ files as input for DFT or post-HF calculations with fine grids and tight convergence criteria for both the optimization and the SCF calculation.
Different input structures will still lead to different outputs, but with smaller differences. It seems you accept that; your argument seems to be that if those differences are <<< 10^-5 angstrom, that doesn’t matter, and I agree somewhat.
I also agree that
so the concept of a minimum point is somewhat meaningless, in reality you have a thermally accessible region of the PES. I still think reproducibility of the exact values is an important and valid concern, but fine, let’s say for now that as long as Avogadro doesn’t stop you from finding your minimum (where minimum means the wider basin rather than a stationary point) it’s not an issue.
However, the differences might not always be so minor, even with tight criteria and accurate numerical evaluation. Even for the best method and algorithm in the world, the gradients calculated for two different input structures will be different, will give different trajectories, and might lead to different minima.
Is that an incorrect statement?
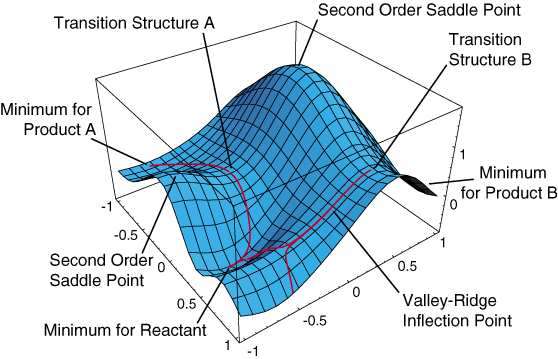
Using this diagram (source) to demonstrate hopefully a bit better what I have been trying to get across this whole time:
If the starting structure in question is very close to the ridge that forms part of the watershed-like red line leading down from TS B, tiny differences in the initial gradient could lead to the optimization descending on different sides of the saddle point into different minima. Even if the gradients are calculated perfectly this remains a theoretical possibility, right?
Maybe a counter-argument would be that the chances of such a watershed lying between two points that differ by max. 5e-6 angstrom in each coordinate is small. But given the complexity and number of dimensions that a PES actually has, that seems to me to mean that it’s a lot of watersheds that we’re hoping to avoid.
What I find remarkable is that even without fishing for specific examples and trying multiple different input geometries, one of the 8 examples I tested already coincidentally showed this exact behaviour. Depending on the input geometry, I got different rotamers of the acetyl group. Sure, this probably occurs more with xtb than with other things, but the point is that it can occur. And there’s no reason why the results can’t differ a lot more than simply being rotamers.
You could say that it demonstrates the importance of doing a conformer search. I agree, it does, but I also don’t think that means it’s not an issue. Imagine the following scenario.
A second scenario:
The outcome in both scenarios is that the use of Avogadro in an intermediate step has directly affected the qualitative results of a calculation purely through overly zealous rounding. If the user then asks a colleague to check what they’ve done or to help them and the colleague tries to reproduce their work, suddenly Avogadro’s implementation of file export becomes relevant.
You might dismiss these scenarios as unlikely, but are you telling me they’re impossible? What is certain is that the less Avogadro rounds off data it imports, the smaller the differences from the original file and the less likely these things are to occur.
Am I actually wrong here? Naturally if my understanding of the chemistry/physics is flawed, I would definitely like to be told so.
As I said, the same xtb calculation repeated 20 times gives identical output to 14 decimal places. Am I misunderstanding what you mean by numerical noise? Shouldn’t noise be random rather than deterministic?
This to me seems not to be about numerical noise, this is not about the inherent inaccuracy of these calculations, this is about the fact that different sets of input values lead to different results even when the differences are at a meaningless level of precision.
Do people regularly use Avogadro’s export function to export such vast ensembles? I can’t actually figure out how I would export a multi-structure XYZ, or a set of single-structure XYZ files, from Avogadro.
For exported XYZ files only, though. Which are far fewer in number than all the CJSON files produced, you would imagine. Maybe I’m getting close to whataboutism here, but CJSON is much bigger than XYZ, which is tiny, it could barely be smaller. For a small molecule like aspartic acid (naturally many molecules are bigger) the CJSON is 4.2 kiB and the XYZ is 627 bytes. Obviously XYZ is far too sparse for use as a save format, so CJSON has to be bigger, but in CJSON we have redundancies like listing the bond order for all bonds rather than just which bonds are double bonds (so on average at least half a byte per bond), or writing the layer of every single atom to file even if everything is in layer 0 (probably one byte per atom), or writing the formal charges even if they’re all 0 (ditto), or writing each display type as
"Licorice": [
false
],
and not "Licorice": false (216 bytes per file). Why indent CJSON by default instead of just printing it all on one line, like some of the CJSONs in the bundled libraries (alone for the atomic coordinates array this costs 24 bytes per atom)? So, and I mean this very politely, why is a 9 byte per atom (8 dp) to 15 byte per atom (10 dp) increase in size of an already extremely minimal format such a huge deal? I just don’t understand.
If it is a significant concern, why are the ORCA and xtb developers so irresponsible as to write the full available precision to every XYZ file they produce? I’d suggest ORCA has written far far more XYZ files than Avogadro.
The size of the molecules and fragments libraries that we ship with every copy of Avogadro are to the tune of 20 MB (times 2 million installs). That 20 MB alone is equivalent to using 10 instead of 5 decimal places for over a million atoms in XYZ files. The crystals library is another 20 MB.
If you are keen to actually do this it’s very much possible with the current library. Trailing zeroes aren’t written when serializing, so all you have to do is round the internal doubles of anything the user inserts or moves to 10 or 8 or 5 decimal places. They are written with full precision because Avogadro stores floats with that many significant figures.
Clearly ORCA uses 64-bit floats. On modern processors I understand the performance gain from using 32-bit floats for calculations to be relatively small, though higher when running calculations on a GPU. But again, if that extra precision is completely useless and has zero effect on anything, why do they not use 32-bit floats for the performance benefit?
By the way, I really don’t see this as making much difference to the theoretical discussion, but as I am assuming that this was intended as dismissal of the two outcomes I found as being merely unphysical artifacts: Would a negative frequency of around -150 cm-1 count as a small imaginary frequency to you? The stationary point in question was a qualitatively different rotamer (trans vs cis ester), so it wasn’t just some artifact.
Anyway, I’ve invested far too much time into this. Realistically it’s probably also a waste of your time to respond to me in full when there’s so much else to do. If you don’t agree with what I’ve written then I’ll just drop it. If you think I’m wrong, that the above is scientifically unsound reasoning, then they are indeed truly useless bytes, this shouldn’t be implemented, and I won’t be opening a pull request. I am not a professor and nor am I an expert in quantum chemistry, so while to me it seemed obvious that these differences are enough to change the outcome of e.g. a geometry optimization in the way that I described, it’s entirely possible that my understanding is flawed.
If you’re interested this can be seen fairly easily by drawing a simple molecule, saving it to CJSON (the coordinates will have 14+ decimal places), then opening the coordinate editor which rounds off the coordinates, then saving it to CJSON again. ![]()
Look, this has clearly wasted a lot of time. And I respect the points of both you and @brockdyer03 as well as your contributions.
I have no problem with a pull request as I’ve said a few times in this thread. I’m just indicating that the choice of 5 decimal places was intentional, and I don’t particularly think an increase in precision buys you much. ![]() That’s why it’s also the default precision in Open Babel.
That’s why it’s also the default precision in Open Babel.
I mention “numeric noise” not that a program run multiple times won’t end up with the same result (it should) but that a small change in compiler or math library, etc. can often change results in the X significant digit because you’re only converging to a certain limit.
In other words, if I run a calculation on Linux and on a Mac, I may very well end up with slightly different energies because x86 and arm64 have different math implementations.
A negative frequency of -150 cm-1 is borderline. And I appreciate the examples. Honestly, it makes me question the GFN2 potential energy surface, but that’s a different matter.
Great. If you’d like to help with a patch to round CJSON to 8 or 10 decimals, that would be great. I fully intend to decrease the size of CJSON as we approach release. For now, it’s indenting because it’s easier to debug. ![]()
As far as other inefficiencies in CJSON… I welcome those too. It’s a community format.
Just so this thread doesn’t go even further - it seems like y’all want an increased precision. Sure. Please submit the patch. And if you’ve got ideas on saving space in CJSON (or elsewhere in Avogadro), I look forward to that too.
To me, it is a matter who uses Avogadro2 and for what purpose. Two cases come to mind (there might be more):
eventually the visualization of a small molecule; a brief / affordable optimization that shows the structure “not too much wrong” after the construction of a molecule with the sketcher and fragments, knowing a force field like MMFF96 doesn’t consider all atom types nor elements; perhaps share/submission to a computational chemist/physicist for an ongoing computation at higher level of theory.
For this, I’m fine with 4 or 5 decimals, the later the default generating / translating a structure as .xyz by openbabel. If one checks e.g., with Jmol’s bridge to Gaussian, or its native export of structures fetched from NIH (with load $benzene; and subsequent write benzene.xyz; on the program’s console). To me, Avogadro is set up fine for this:
Avogadro is used to read the results of Gaussian, MOPAC, etc. and these results with atomic coordinates of say 10 decimals in Angstom or Bohr are to be reused for an optimization at higher level of theory, or/and computation of a property previously not considered. Then, it probably could be a loss if not all decimals are carried over to “the next round”. To me, this looks to me like Avogadro is requested to transfer strings from one into the next file – different from optimizing for instance a bond angle by Avogadro with its limited tools.* Then, it would make sense if Avogadro read the log of Gaussian, and will provide an export of the xyz coordinates with “Gaussian precision”**, too.
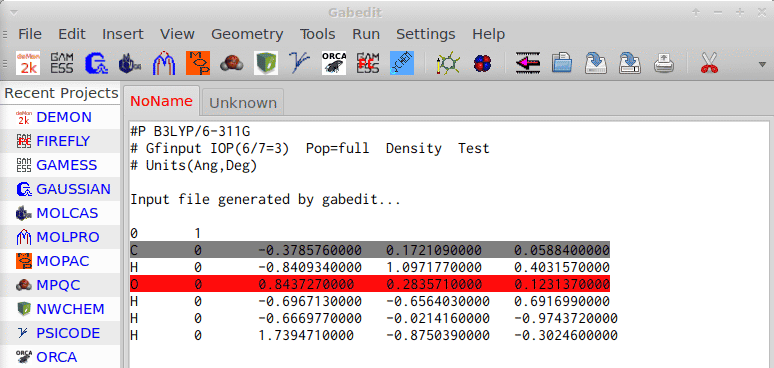
To me, this incremental use of better and better theories to get the result eventually wanted is the/one reason why after performing an initial geometry optimization of methanol with Amber potential im gabedit provides 10 decimals; and not that the many zeroes (already) contain (chemical) information:
* I don’t think the Avogadro project wants to build an additional Gaussian, GAMESS, etc; and to me, it doesn’t need it either.
** Contrasting to e.g. Openbabel which provided a GAMESS output (coordinates in Angstrom, 10 decimals) reports the .xyz rounded to 5 decimals.