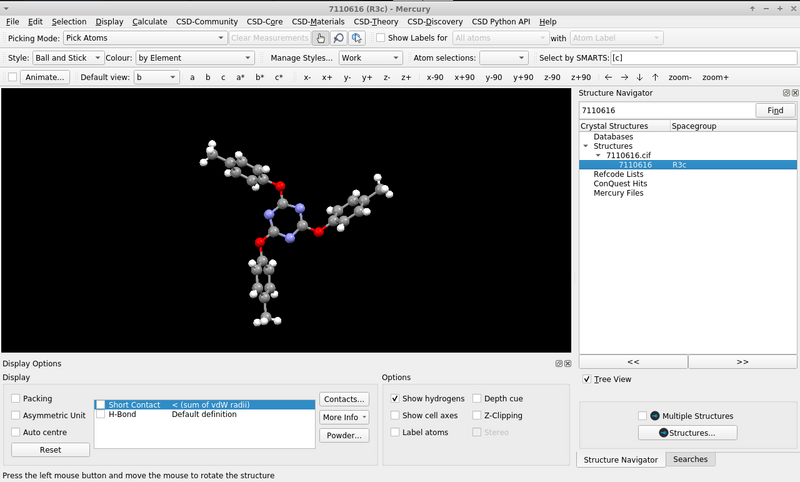
I presume Jmol and JSmol share the commands the user can issue. If one reads the .cif locally (on the program’s console e.g., load "7110616.cif";)* the molecule is presented in the “symmetry completed” form:
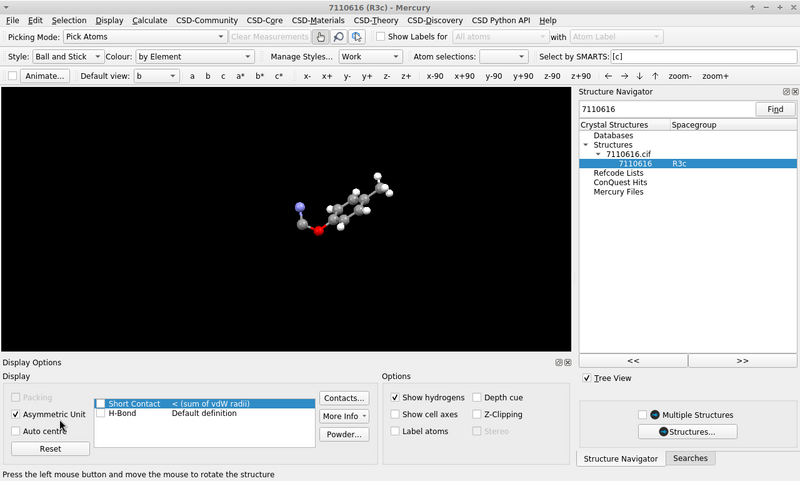
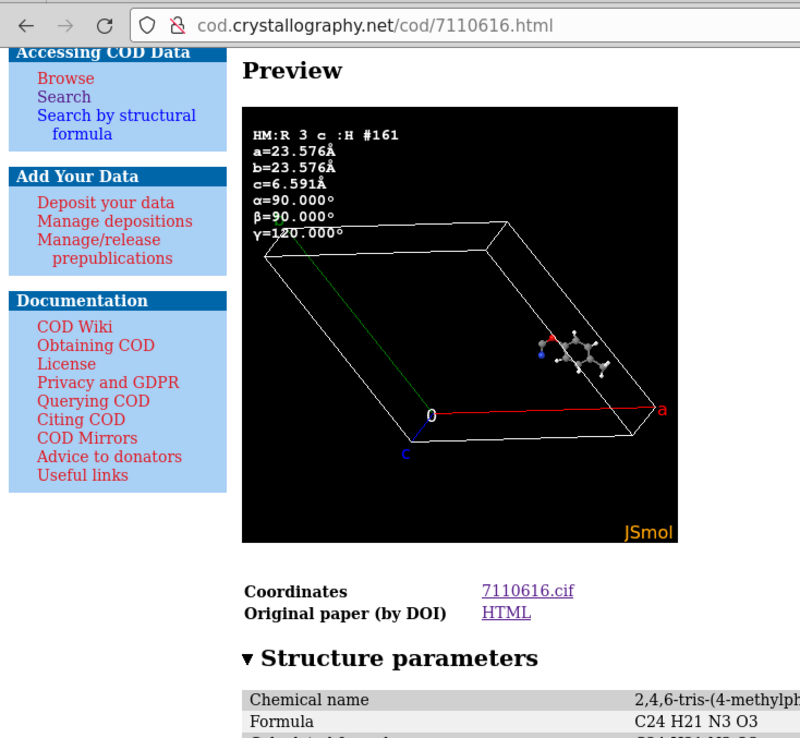
The visualization by COD with JSmol is the asymmetric unit, because it runs the command load "7110616.cif" {1 1 1}; hide symmetry; instead.
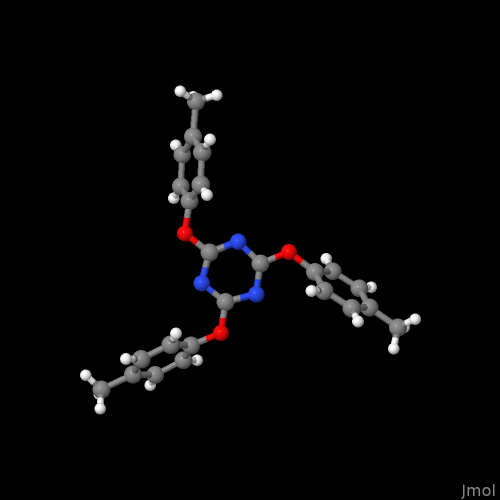
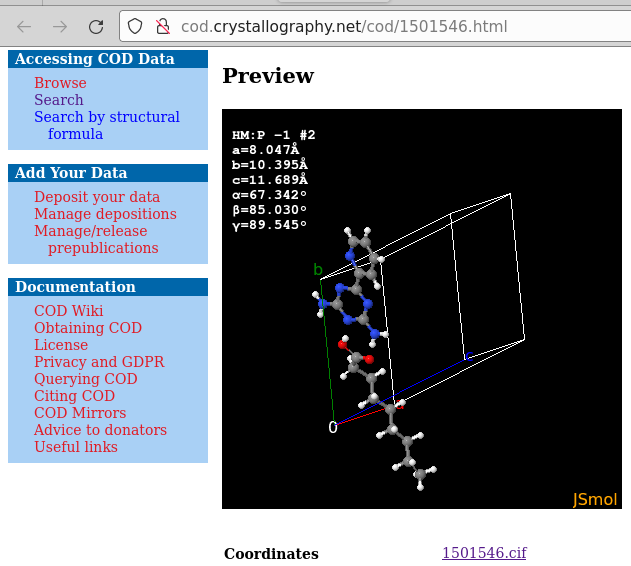
Entry _chemical_formula_moiety in a cif file is a bit more complex. It reports with individual Hill formulae (charges permitted) discrete entities e.g., separate ions, solvent molecules (e.g., hydrates), “the other component” in accidental / intentional co-crystals. See e.g., COD 1501546 with the lines
_chemical_formula_moiety 'C9 H18 O2, C8 H8 N6'
_chemical_formula_sum 'C17 H26 N6 O2'
Here, though P-1 is a space group with centre of inversion, there is no “completion of molecules by symmetry”, each position in the asymmetric unit was defined in the atom block:
But it isn’t always present e.g., in COD 1010527 about \ce{CuSO4 * 5 H2O} reading
_chemical_formula_structural 'Cu S O4 (H2 O)5'
_chemical_formula_sum 'Cu H10 O9 S'
I speculate the maintainers of the COD might help to identify an implementation suitable for Avogadro (e.g., Andrius Merkys, Antanas Vaitkus). Their CLI tool codcif2sdf in cod-tools (which is packaged for DebiChem) has to tackle a similar task to join atoms back into a format “pure chemists” recognize easier. It works really well (which includes the assignment of bond orders) for plain organic structures. Its action is limited either by the chemistry (bond/bond orders in organometallics / inorganic compounds), or the structure model (models with positional, or occupational disorder are better checked/edited manually).
* With a ~/.bashrc amended by an
alias jm="java -jar ~/.../Jmol.jar -L"
the first time a structure file is load into Jmol only requires a jm 7110616.cif. Beside the tab completion by the shell, this retains access to a set of useful flags jm -h lists.