Would anyone kindly share, once downloading a python generator, how to set python path to generate and read-in gaussian files? Thank you in advance!
I think you’ll need to be more specific as to the operating system you’re using and how / where you’ve installed Python.
-
In case you access a Linux of Debian (or a relative, like Ubuntu/Xubuntu), Extensions → Set Python Path is the first step to go
If you do not know where your Python interpreter is, the command
which pythonguides you. Depending on the installation at your disposition, it may be namedpython3(like above), especially (although not limited to) installations which (still) know/knew legacy Python2. In other instances (e.g., a pristine Ubuntu 20.04 LTS and above),pythonrefers by default topython3. So run the quick complementary more explicit checkswhich python2andwhich python3to ascertain you indicate Avogadro the path of Python 3. -
As second step, go Extensions → Download Plugins → Avogenerators to get the exporter bridges working. In case of Debian, if an earlier version of Avogadro was used (either as AppImage, or installed as a .deb package), it may be helpful to clear and remove all old content from path
~/.local/share/OpenChemistry/Avogadro/inputGenerators
including the very folder inputGenerators itself before this download and automatic extraction of the required files starts. Eventually, this writes a structure like
~/.local/share/OpenChemistry/Avogadro$ tree
.
└── inputGenerators
└── avogenerators
├── dalton.py
├── gamessuk.py
├── gaussian.py
├── generators.cmake
├── inputgeneratortest.py
├── LICENSE
├── molpro.py
├── mopac.py
├── nwchem.py
├── orca.py
├── plugin.json
├── psi4.py
├── pyscf.py
├── qchem.py
├── README.md
└── terachem.py
2 directories, 16 files
After restarting Avogadro and drawing a structure, the GUI’s entry input then offers 16 formats (including position 8, Gaussian).
Thank you - I am using windows 11 pro; Python is installed on my local hard drive (C-drive) - same drive for operation system and Avogadro.
It seems as if Conda / Miniconda does not add Python to your PATH on Windows anymore.
As @Thomas mentioned, you can go to Extensions => Set Python Path
I would guess something like C:\ProgramData\Miniconda3 if you installed for “all users” or C:\Users\MY_USERNAME\AppData\Local\Continuum\miniconda3 (where MY_USERNAME is your Windows user name).
Thank you - I have set python path for a Window’s OS; I now can generate gaussian’s input files, but I am still unable to read-in gaussian’s “.log”, “.out” files. I also cannot read-in “.mol” files. Any advice here would be greatly appreciated!
Thank you - ghutchis. I don’t believe I have conda/miniconda but I was able to set python for Guassian input files. I am still unable to read-in gaussian’s “.log”, “.out” files. I also cannot read-in “.mol” files. Any advice here would be greatly appreciated!
For the .mol file: do you have an authentic reference file at hand? If not generate one with OpenBabel from the command line, e.g.
obabel -:"c1ccncc1" -h --gen3d -O pyridine.mol
to write one about pyridine. Then (now in Avogadro) access the GUI entries File → Open (or short, Crtl + o) which opens a file manager window. Confirm the question “close without saving”:
Then either select “All supported formats” in the line “Files of type” (it is a pull-down menu):

or narrow down to the MDL mol format:
For the reports written by Gaussian, you need to know which version of the program you use; there were small differences how results are formatted/presented. You find this information very early in the .log/.out written by in plain ASCII when stating the copyrights. For reading them with Avogadro, check if the file extension matches the ones anticipated by the import, e.g. example.g16):
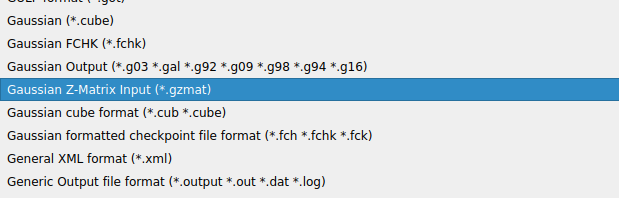
This is necessary because *.out and *.log are popular file extensions not specific to Gaussian. (Indeed, Avogadro may misunderstand a file in pattern of example.out as permanent record written by GAMESS or NWChem, which are other programs in the field.)
In case you need to familiarize yourself with these file formats, Jmol has compiled a brief description of some which are more frequently met, including an entry for Gaussian and small public reference files (entry Gaussian log/out files). Thus, after adjusting the file extensions, the examples H2O.out and H2O_3.log are read and visualized well e.g., by Avogadro 1.97.0.
@Thomas here a hyperlink to a “.mol” file I am working with Microsoft OneDrive - Access files anywhere. Create docs with free Office Online.
I’m not sure what exactly your roadblock is. By syntax, the .mol file you shared is fine; however ChemDraw’s .mol files exported use implicit hydrogen atoms, (sadly still) are 2D objects (the z-coordinates are all zero), and without running an affordable forcefield geometry optimization (UFF, MMFF94, etc.). This is why their import in Avogadro – though accepted by syntax – yields the many indications of “short contacts”:
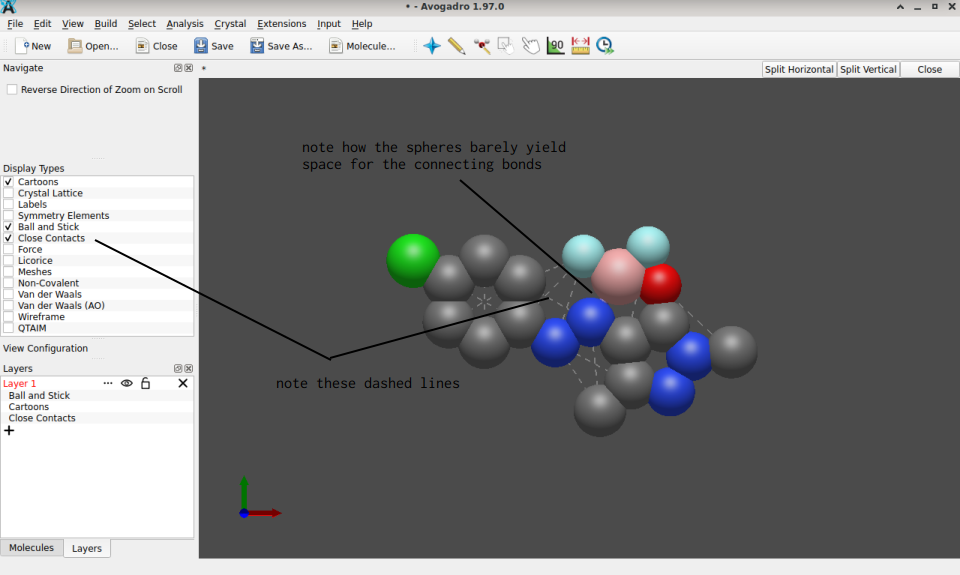
On occasion, after loading the mol file in question, I have to use click (left mouse boutton) once on the blue penrose (top row) or/and the canvas to complete Avogadro’s reading; it possibly is a local issue with my installation/setup.
The missing explicit hydrogens however is something the inspection of the .mol with any text editor may reveal quickly (e.g., Notepad++, or note a compilation by wikipedia). Thus I would recommend to improve the .mol file in first instance. Either a) outside Avogadro by help by openbabel on the command line with a pattern like
$ obabel BF2-OxyBorole-4Ph_MeAAP.mol --gen3d -h -O with_hydrogens.mol
which yields a file like the one pasted on pastebin. (If you want to get familiar with the underlying syntax, get in touch with the public documentation which is online [section of typical examples of instruction], and equally may be downloaded e.g., as as a .pdf.)
Or b), Avogadro’s GUI has the entries Extensions → OpenBabel → Add Hydrogens, Extensions → OpenBabel → Optimize Geometry to provide the same result by mouse clicking.
Eventually though, both approaches yield a structure more suitable for further computation:
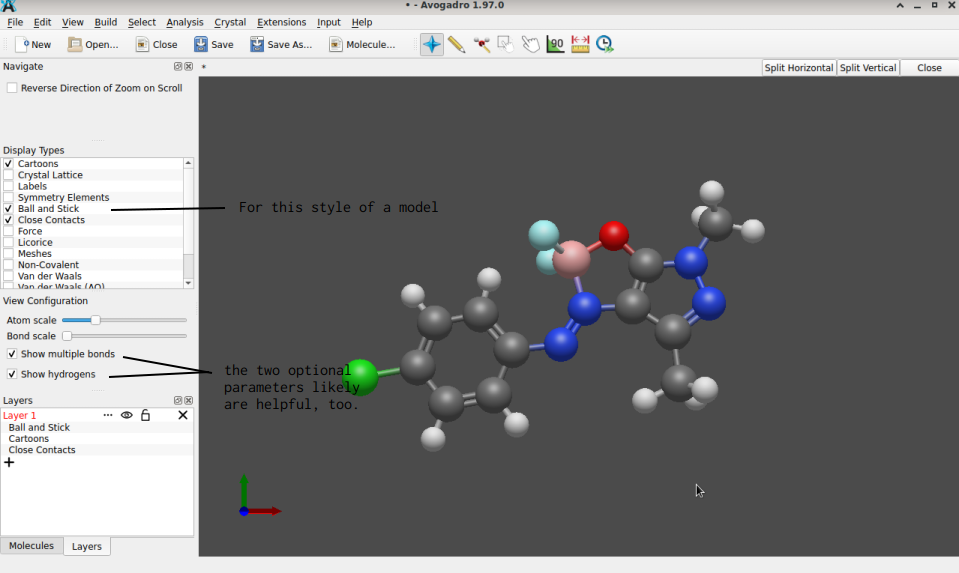
(The Jmol index mentioned earlier equally has an entry with reference files of the .mol format, too.)
Thanks both @ghutchis and @Thomas - you two have helped me in the last two days than all my efforts this summer. I was able to navigate around adding hydrogens and optimizing the geometry to generate a normal looking ball and stick model. For some reason, Avogadro crashed with attempting to add hydrogens, but I was able converted the “.mol” to add hydrogens in openbabel, then I optimized the structure from extensions in Avogadro.
Nice to read that the problems were resolved successfully. Now if Avogadro (which release are you using? In the GUI, there is the entry Help → About to state both the version of the program, as well as the qt responsible for the visualization) continues to shut down unexpectedly, maybe free RAM still available to the system (or its lack) might initiate Avogadro’s sudden closures.
Let the process monitor run (there was a tab displaying the working memory) in parallel when you start up Avogadro. Reading the .mol of a small molecule like your oxazaborol shouldn’t consume (too) much in addition. However the automatic addition of hydrogens and subsequent structure optimization (Extensions → OpenBabel → Configure Force Field allows some adjustments for these affordable guesses) becomes more and more demanding by the number of atoms to consider and their number bonds (e.g., flexible C-C single bonds in chains, methyl groups); this would be one plausible cause for the sudden exit. (OpenBabel’s documentation includes a dedicated section about balancing effort invested, and quality of the model suggested.) So sharing these details may help Avogadro’s growth in the longer run.
Thank you @Thomas, but it looks like I spoke too soon. I am unable to save gaussian input files. I get the below error message. Here’s a video of what happens when I try to save gaussian input. → https://1drv.ms/v/s!AgB7mqM1YFV9md0JcBr4BBktBH5_0g?e=I7fOoz
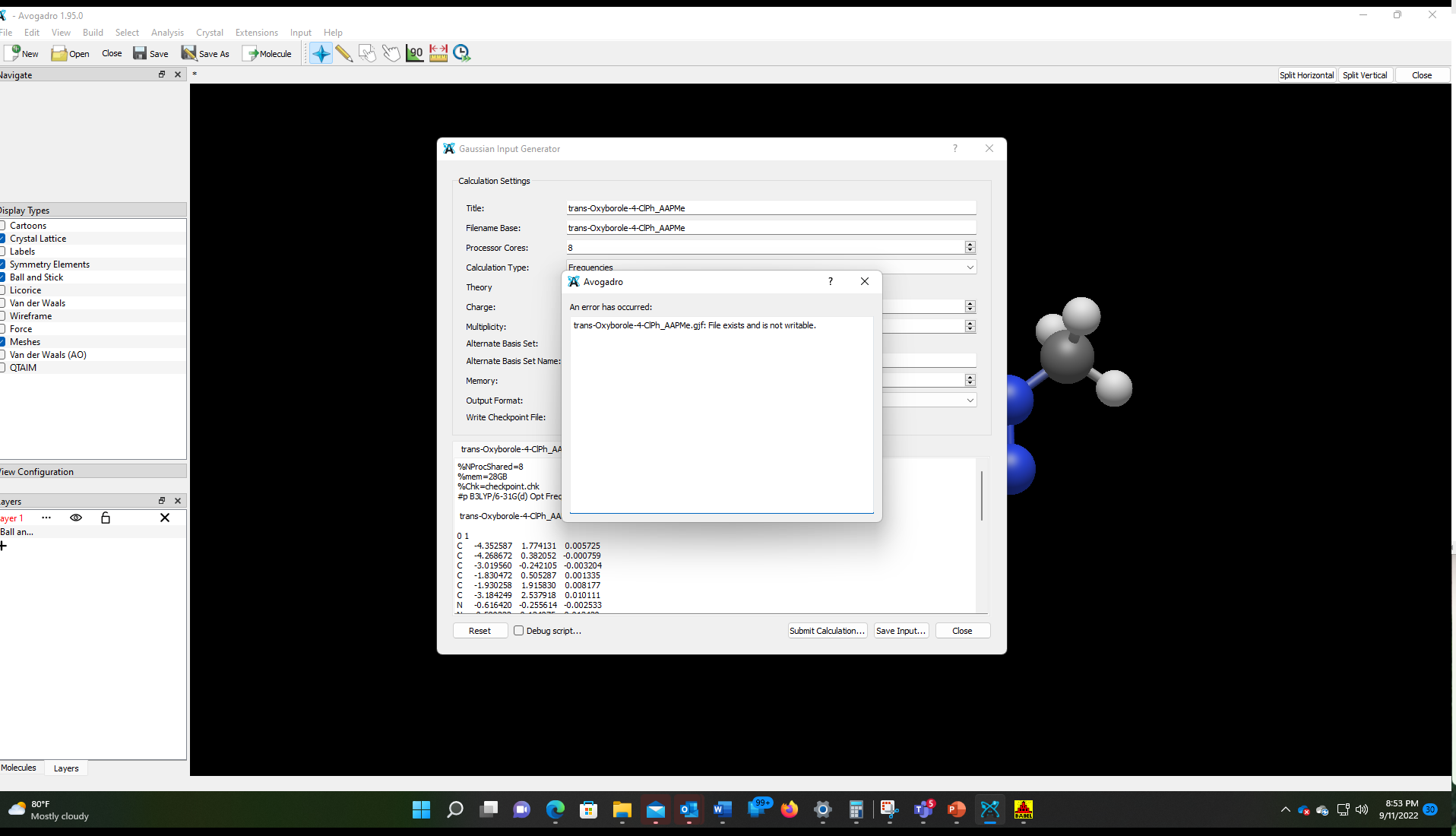
I’m unable to replicate your difficulties. With a recent AppImage for Linux running in Linux Debian, the import of the .mol file, its visualization, and preparing an export for Gaussian did not yield an error I spot:
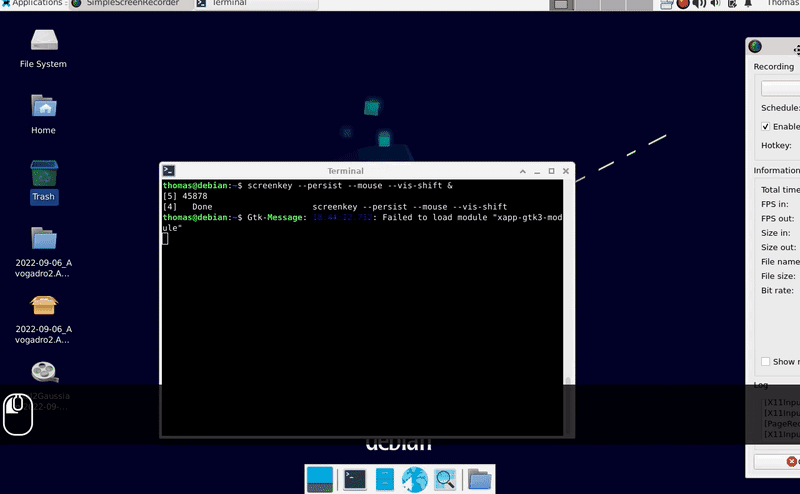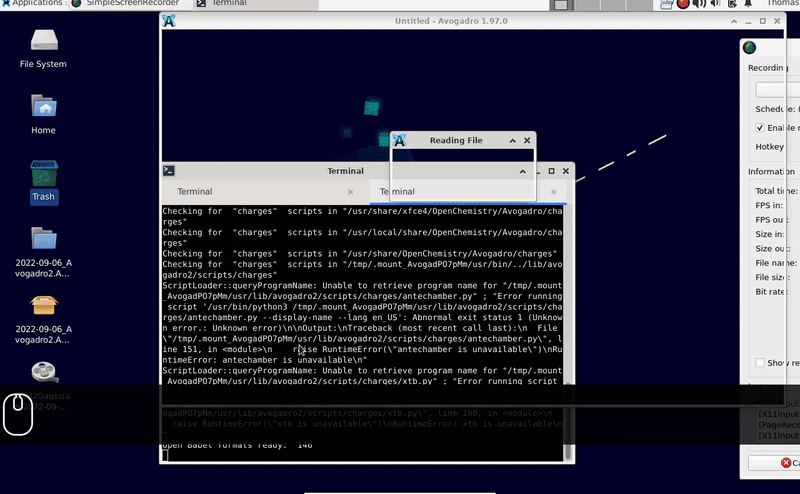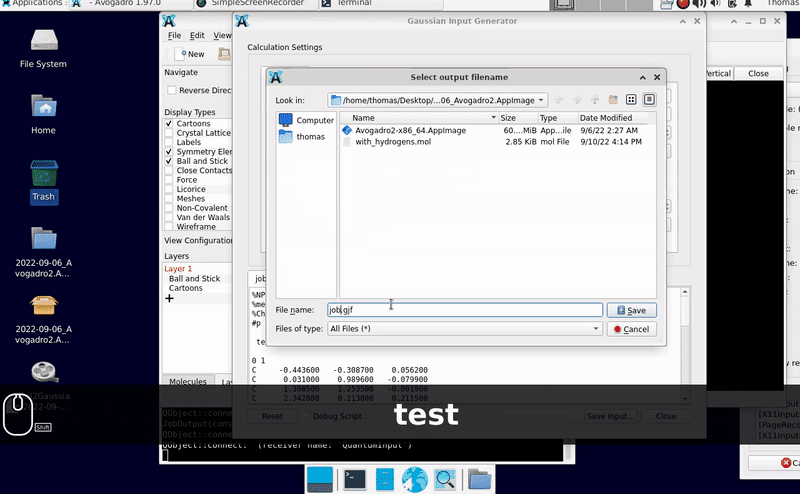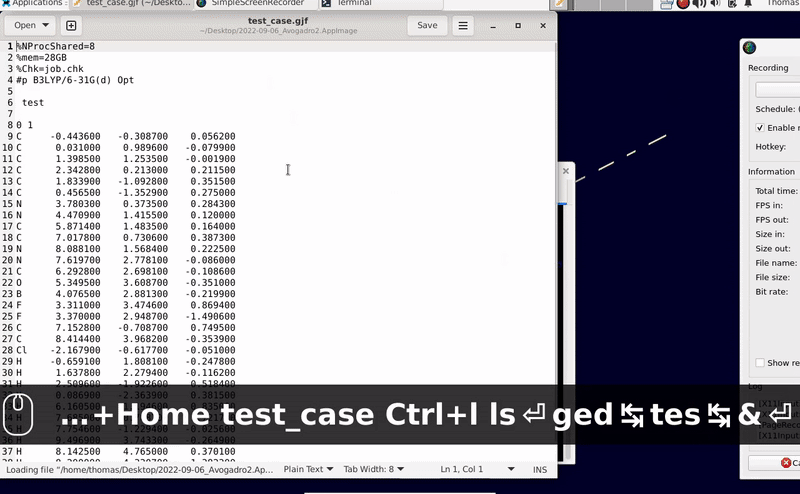
While I have no possibility to check the processing on Windows 11 (perhaps @ghutchis uses it?), maybe you can test a Debian live image running from a USB thumbdrive (or an optical drive) and one AppImage from GitHub for comparison. It is odd that the export into a file does not work though Gaussian’s input already is displayed to the screen … This is puzzling indeed.
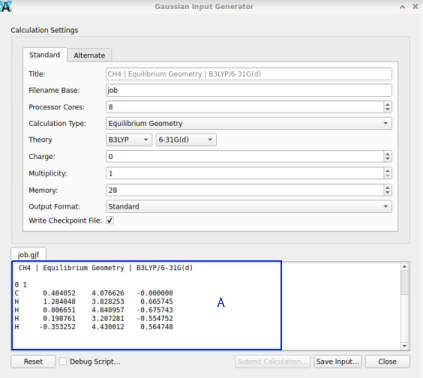
Addition: Should the export from Avogadro fail, one may rescue the content from the lower part from the widget (in the screen photo below the rectangle in blue marked “A”) by copy & paste into a text editor: