I believe this to be a bug with Avogadro / the interface to xtb.
Avogadro version: 1.98.1
A non-zero energy is returned.
An energy of 0 is returned. This also means that, for example, a geometry optimisation using GFN-FF does nothing.
Create a small molecule (e.g., methane), set the force field to GFN-FF. Select calculate energy. Dialogue box returns “ENERGY = GFN-FF 0”
I’ve managed to get a simple energy calculation using the same xtb-python to run via an interactive Python session. Using an alternate FF (e.g., UFF from openbabel) seems to work as expected.
Hmm. Can you try optimizing with GFN-FF? Does that work?
Attempting to optimise with GFN-FF doesn’t appear to do anything visually. I captured the following terminal output:
initial 0 gradNorm: 0
And so on. It seems like Avogadro is not reading the information from XTB correctly?
Hmm, okay. I’ll take a look soon. I guess for you this shows up on any molecule.
It appears that way. I’ve tried building a few different molecules in Avogadro and reading in xyz files with the same result.
In case it helps with tracking down the problem, I’ve tried “running” gfnff.py interactively from a Python prompt using a cjson that I saved from Avogadro. I got the following from the energy print:
print(“AvogadroEnergy:”, res.get_energy())
And the gradient:
output = np.array2string(grad)
A warning, possibly benign, that was generated as I was playing around came from
coordinates[i] = np.fromstring(input())
Which gave:
DeprecationWarning: The binary mode of fromstring is deprecated, as it behaves surprisingly on unicode inputs. Use frombuffer instead
EDIT: changing np.fromstring to np.frombuffer did not help.
You might try this, which gets rid of that warning.
coordinates[i] = np.fromstring(input(), sep=' ')
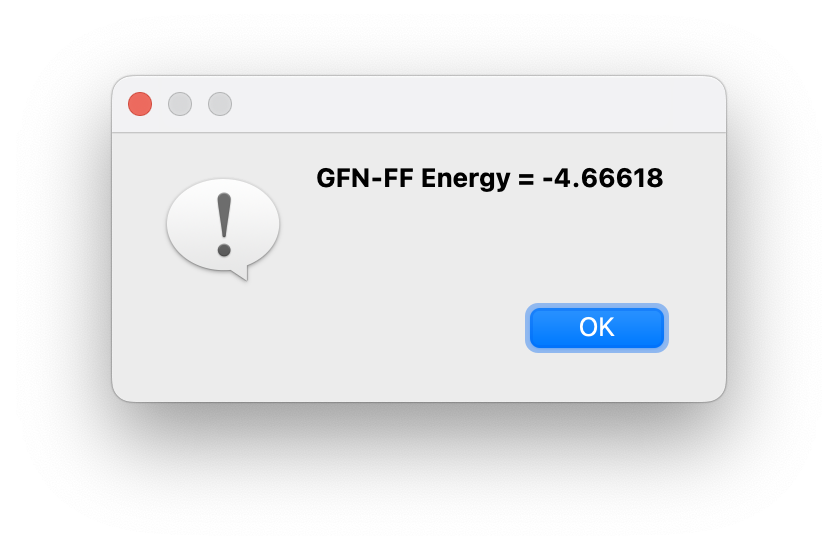
I’m still a bit mystified at the bug. For example, I’m able to successfully optimize caffeine:
maxSteps 250 steps 50
optimize 0 -4.66618 gradNorm: 490.312
optimize 1 -4.67067 gradNorm: 305.669
optimize 2 -4.67761 gradNorm: 31.6003
optimize 3 -4.67783 gradNorm: 21.3193
optimize 4 -4.6779 gradNorm: 18.9278
…
But then as you say, when I ask for the energy command, I get 0.0.
Seems like even though I’ve requested “MUTED” the gfn calculator is giving me output from the SCF calculations:
==================== Thresholds ====================\n CN : 150.00000\n rep : 500.00000\n disp: 2500.00000\n HB1 : 250.00000\n
I’ll tinker with the Python script a bit more. Seems like there are some ways to redirect the output.
Okay, I got it working… (units would be helpful)
I needed to send the GFN-FF messages to a black hole. Feel free to just grab the updated script:
OpenChemistry:master ← ghutchis:fixup-script-energy
opened 08:39PM - 14 Nov 23 UTC
Developer Certificate of Origin
Version 1.1
Copyright (C) 2004, 2006 The Lin… ux Foundation and its contributors.
1 Letterman Drive
Suite D4700
San Francisco, CA, 94129
Everyone is permitted to copy and distribute verbatim copies of this
license document, but changing it is not allowed.
Developer's Certificate of Origin 1.1
By making a contribution to this project, I certify that:
(a) The contribution was created in whole or in part by me and I
have the right to submit it under the open source license
indicated in the file; or
(b) The contribution is based upon previous work that, to the best
of my knowledge, is covered under an appropriate open source
license and I have the right under that license to submit that
work with modifications, whether created in whole or in part
by me, under the same open source license (unless I am
permitted to submit under a different license), as indicated
in the file; or
(c) The contribution was provided directly to me by some other
person who certified (a), (b) or (c) and I have not modified
it.
(d) I understand and agree that this project and the contribution
are public and that a record of the contribution (including all
personal information I submit with it, including my sign-off) is
maintained indefinitely and may be redistributed consistent with
this project or the open source license(s) involved.
I can confirm this works for me - thanks for tracking it down and fixing!