Hello,
I am curious if there is a way to indicate a carbocation in avagadro so that when EEMs (electron density map) are generated, the electron density reflects the electron deficiency. Currently, when I try to generate a carbocation (such as (CH3)3C+) the EEM only reflects the anionic species instead. Please let me know if there is a workaround for this.
Thanks!
I’m not entirely sure I understand. You’re using the EEM charge model in Avogadro 2? Sure. That’s going to reflect neutral molecules. (So I’d be surprised if the charges for a carbocation reflect an anion.. EEM was designed for neutral species.)
If you really want an electrostatic map for a charged species, I suggest using the XTB plugin for example – it will run a quick approximate quantum calculation using GFN2 which would take into account the charged species.
1 Like
Thank you for your response! I was able to get the xtb pluggin working for avogadro but this may be a dumb question. How do I run the quantum calculation to account for charge species, and how do I indicate a charged species. Thereafter, how do I generate an EPM based on these calculations. Any help is much appreciated.
Under Analyze > Properties > Molecular you can set the overall charge on the molecule, and the xtb plugin will pass it through to the xtb calculation.
1 Like
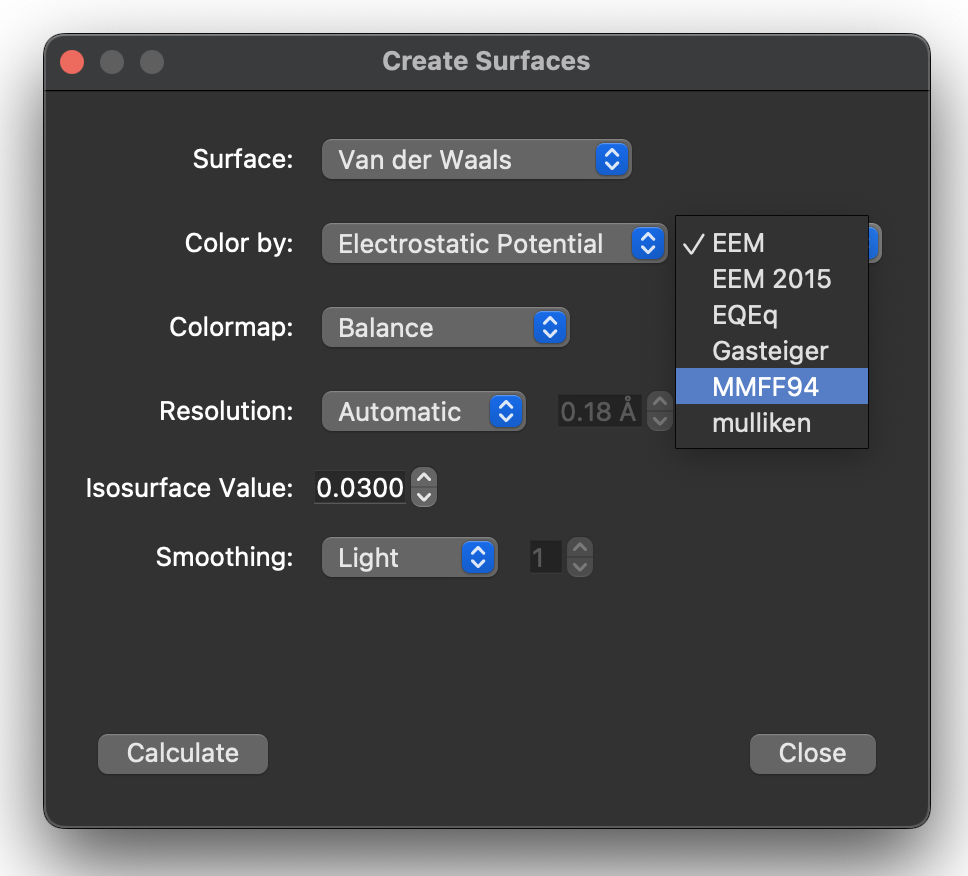
Thank you I was able to set the overall charge on the molecule. However, how do I generate the EPM using the xtb calculation. I only see the pre-set options (EEM, etc.) when trying generate the molecular surface and it still outputs electrostatic potential maps that do not take into account the specified charge.
Thank you!
Let me take a look - it should be reading in the charges from the xtb calculation, but there may be a bug either in Avogadro or the plugin.
The alternative is for me to post the GFN2 charge plugin, which runs xtb from your path and reads the charges directly. I’ll see if I can put that up today.
Thank you, the GFN2 charge plugin would be great
Yeah - the delay has been that the current code doesn’t set the total charge for the GFN2 calculation. (And the usual mid-semester chaos)
I’ll have a build set up soon - it’s clearly a useful request.
I’m also trying to figure out why the charges aren’t read back after using the current xtb plugin.
Okay, it looks like the xtb plugin works already, but you have to scroll down to find the mulliken charges:
(Note to @matterhorn103 - these are probably better listed as GFN2 charges, but that’s a minor thing.)
I have to make some patches to Avogadro to get the script working directly with charged species. (At the moment, it uses a mol file which doesn’t include the total charge on the molecule.)
I’ll post here when I have a patch later this week.
Ah, I didn’t realise that the JSON key would be showing up in the interface in that way. I can change it, no worries, the origin of the values should definitely be made clear.
It seems important to me though to note that they are Mulliken charges, since it’s otherwise a bit of an effort to find that out, and xtb does have a separate electrostatic potential runtype that might be useful to add to the plugin later (though I don’t know to what extent that calculation differs from the one Avogadro does). So I’ll probably list them as Mulliken (GFN2-xTB), if you agree that’s reasonable?
Glad to hear that the plugin works though! @Isaiah_Nelsen –just note that some runtypes don’t return Mulliken charges, but an Energy calculation definitely does. So you might in some cases have to run an extra Energy command, after something like a Frequencies calculation or a Conformers run with CREST.
1 Like