Avogadro version: 1.99.0 Nightly Build 11/4/2024
Operating system and version: Windows 11 Pro Version 10.0.22631
Expected Behavior
Molecules with a formal charge should have a formal charge.
Actual Behavior
All formal charges are zero.
Steps to Reproduce
I have tested [1,2,3]-1,3H-triazolium (SMILES c1[nH+]n[nH]c1) and hexafluorophosphate (PF6-), and no formal charges are stored, all the charges are zeros. I have yet to attempt to generalize this to all molecules, but that sounds like it would take an amount of time greater than the lifespan of the universe, so I am going to put off on that until I finish my thesis.
Edit
I think I might have an idea of what is happening. In chargemanager.cpp, there is a check for whether or not partial charges exist, but the partial charges are initialized in molecule.cpp and so there already exists a bunch of zeros in the cjson where the partial charges should be. I am not 100% familiar with the way that the detection method works, but it doesn’t seem to detect what the value is, just if it exists. Perhaps checking if all the charges are zero should trigger a calculation of them again, and then set some thing to freeze the check until perhaps the molecule changes?
How did you build the molecules? And when you say “no formal charges are stored” … what does that mean?
I’m not sure I understand. You mention “all formal charges are zero” but then you’re talking about partial charges? The chargemanager.cpp code handles partial charges, not formal charges.
Did you build from a SMILES and are wondering why the formal charges from the SMILES are not retained?
Did you manually build a four-bond nitrogen or a 6-bond phosphorus and wonder why the formal charges are not automatically perceived?
It might help if you could briefly explain what Avogadro now attempts to recognize/compute when it comes to charges and spins, I’d also be interested.
But my understanding is that partial charges are intended by you to be a property that is calculated elsewhere (including potentially by plugins) and displayed by Avogadro, not something that Avo should attempt to automatically allocate. And that the recent work you did was instead about automatically assigning an overall charge and spin to the whole molecule based on valencies.
By the way, I was checking out the new molecular properties work myself and I noticed two bugs:
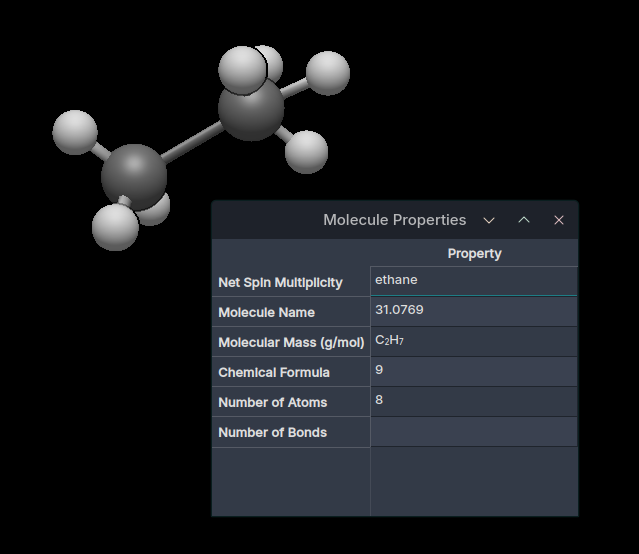
If I draw a normal neutral molecule and go to Molecular Properties, the multiplicity and charge are not shown (I don’t mean just that the numbers are missing, I mean that the respective rows are missing entirely)
If I make a molecule hypo- or hyper-valent, a key for Net Spin Multiplicity appears, but it appears in the wrong place with the wrong value; it is matched with the value for Molecule Name and everything is shifted. And the value for the multiplicity isn’t shown at all anyway. And charge still isn’t shown. See the screenshot:
So it seems the automatic calculation is working fine, just not the display. Though I’ll note that the automatic calculation of the multiplicity was already working the same for this molecule in the AppImage from three weeks ago, so I guess that wasn’t added by your recent work.
I tried 5 methods, one was using the ‘Insert SMILES’ option (triazolium), one was using the ‘Download by Name’ option (PF6), one was from importing an XYZ file (PF6), one was just building the structure by hand (triazolium), and the last was reading in an output from an ORCA calculation (not sure that even is supposed to work, just was being thorough).
I think I mean both? I am not 100% sure, since I saw that the chargemanager.cpp file did partial charges, but in the cjson there is a section labeled formalCharges, I assumed that they may have been the same thing.
I indeed did make these molecules and wonder why the charges are not perceived or calculated somewhere along the way. There doesn’t seem to be any case where the formal charge part of the cjson is not all zeros, which seems like it may be a bug. In addition, due to the fact that the totalCharge is listed as zero no matter what, the spin multiplicity that is calculated is incorrect with things that have a non-zero charge.
No, they are absolutely not the same thing. Personally, I’d love to see formal charges banished from chemistry because students have confusion when some atom doesn’t actually have a partial charge anywhere near +1 or -1. (Even for something like \ce{Na+} or \ce{F-}.
If you haven’t specified any formal charges, the total charge will be zero.
I do know that formal charges and partial charges are different, and I know formal charge isn’t real (I have read many many papers on the topic for research). I was simply making an assumption regarding the way that the things are listed in a .cjson file.
I forgot there was a way to specify formal charges, and in my search for where that was I stumbled across something interesting. Formal charges are in the atom properties box, but not if you copy and paste the molecule (which is akin to copy pasting a cjson I believe)
Further, the formal charges ARE stored if you export the molecule as a .cjson file. There seems to be some thing that isn’t updating the active molecule’s information, but is indeed calculating the formal charge and feeding it somewhere.
Edit: The formal charges are not saved when exporting a .cjson from an ORCA output, and there are no partial charges either.
I’m not sure I understand. You opened an ORCA output file, assigned formal charges, but they’re not saved to cjson? Or are you expecting that formal charges would be perceived from the output file?
I don’t remember if the ORCA input reader currently grabs the partial charges from the output file.
I opened an ORCA output (attached below) and I noticed some very strange stuff. In no particular order,
Formal charges are calculated nowhere, not the atom properties, not in the active cjson, and not even if I export as a .cjson
The partial charges in the atom properties menu do not match the ORCA output, I can’t even tell what they are from, but I do know that they add up to zero when they should add up to -1
The total charge is not correct anywhere in Avogadro