Avogadro version: Avogadro 2 (1.99.0) Nightly build as of 6/17/2024
Operating system and version: Windows 11 Pro Version 10.0.22631
Expected Behavior
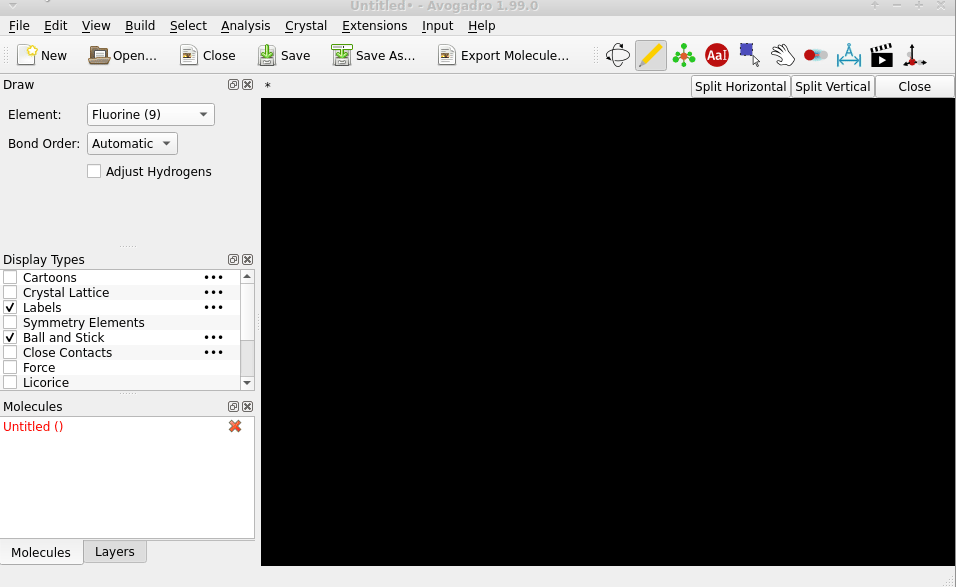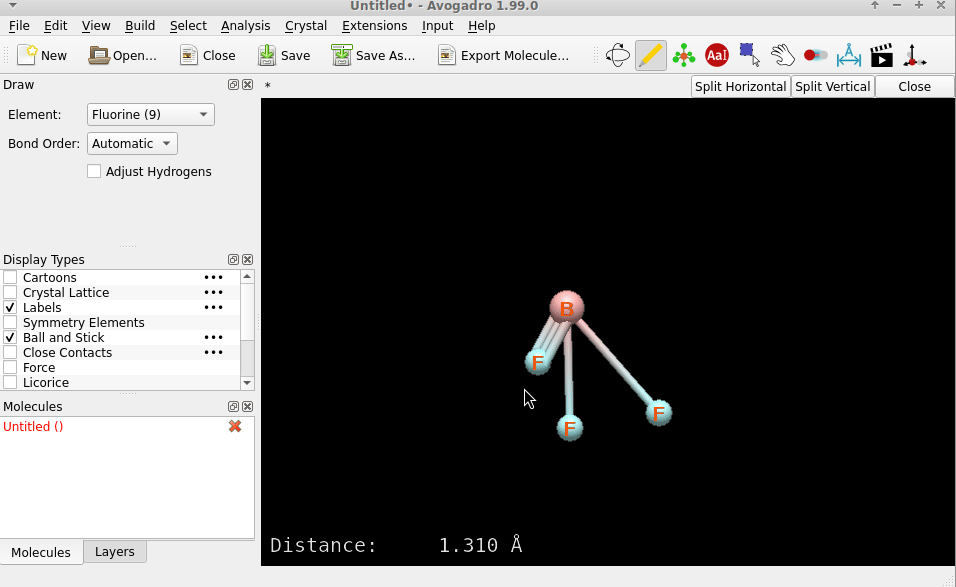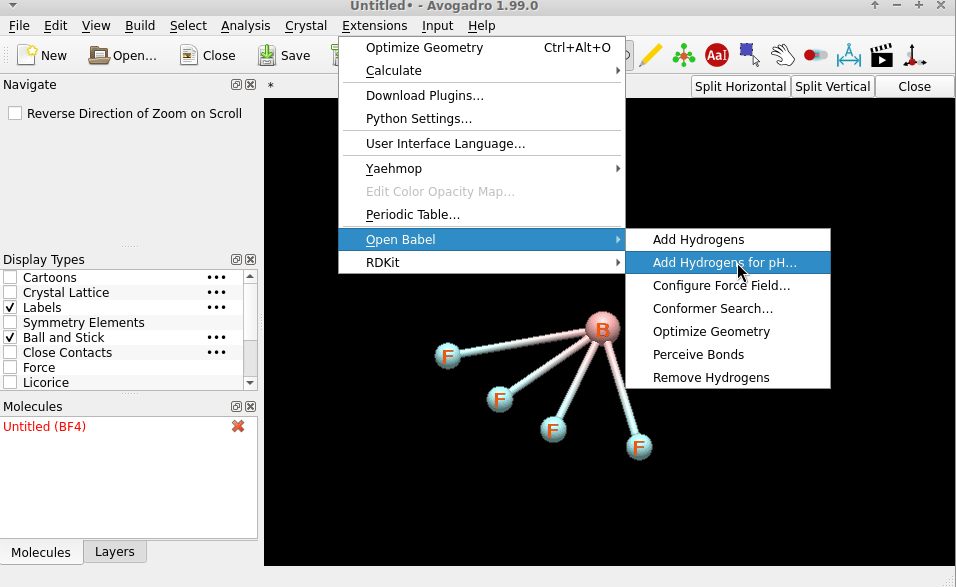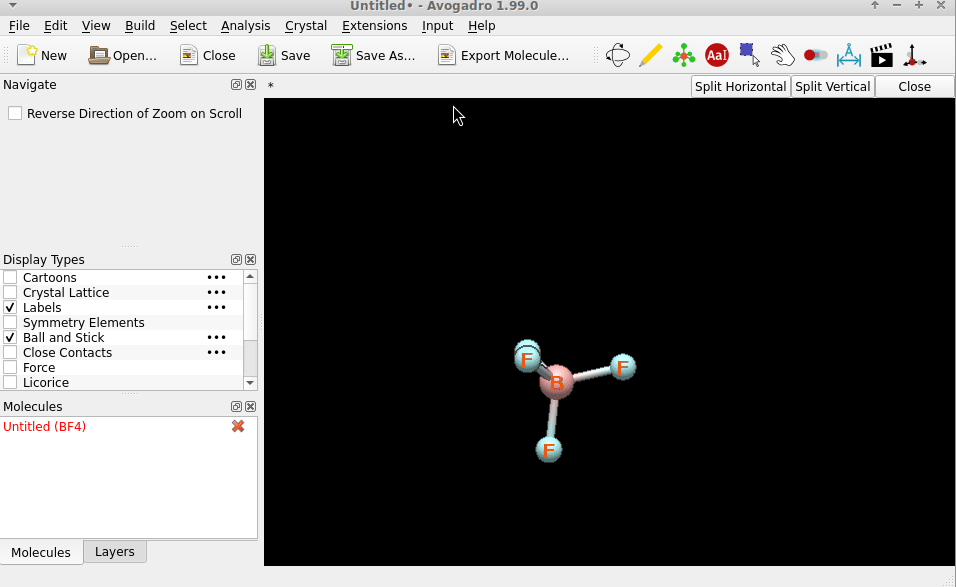
Navigating to Extensions>Calculate>Configure>Optimization Method should allow the user to select a force field.
Actual Behavior
The option to select a force field from the drop-down menu is grayed out, and can’t be selected
Steps to Reproduce
This issue was noticed specifically with using borates, (BR4)-, with R = Any, but the issue is not exclusive to this kind of molecule. To reproduce, draw a molecule such as tetrafluoroborate, and attempt to navigate to the menu mentioned above. The menu drop down should be grayed out and should be unable to be selected.
Possible Ideas
It seems that when a force field is not detected automatically the program seems to just default to disabling all choices of force field, even though one of them may work. I suspect that somewhere in the code for picking a force field there is something like a boolean that gets set to false and that same boolean controls the availability of the force field drop down menu, or perhaps controls which force fields are available in that menu.
I suggest that if this is the case that the force field menu should constantly be populated with all of the default methods that AV2 allows, such as GAFF, GFN1, GFN2, GFNFF, MMFF94, and UFF. Allowing selection of any of these even when it may be an inappropriate choice would save the user from getting stuck with just the OpenBabel implemented fields.
@brockdyer03 Do you have access to an instance of Linux? I just drew a neutral BF4 molecule in Avogadro 1.99 (Linux AppImage, fetched 18th May) in an instance of Linux Debian 13/trixie (which by virtue of this operating system already has OpenBabel available as a package in Debian’s repositories). Here, an intentionally skewed input structure was optimized by UFF into a more reasonable tetrahedral geometry:
I presume there would be a similar success in Linux Ubuntu, too. On the other hand, MMFF94 set up to cover typical small organic molecules (of pharmaceutical interest for Merck in Rahway/NJ) likely didn’t (yet) considered \mathrm{BF^-_4} (reference) and similar compounds.*
You might give some additional ropes here, I interpret your report like a suggestion Avogadro should recognize e.g., by elements/atom type present (and hence guide its user) if one of the force fields generally available (e.g., by OpenBabel) has the parametrization to process the specific structure “well enough”. This would pass beyond
* Thinking e.g. about some coupling reactions by the Gary Molander Group/University of Pennsylvania is known for.
The OpenBabel implementation works fine, I am not incapable of using that feature to optimize structures, what I am reporting here is that the native implementation of force fields is bugged.
The code will populate the menu on the basis of the elements, etc. For example, it’s not possible to perform a MMFF94 or GAFF calculation with boron. So no, I don’t think it should show those options.
UFF should be available, as should GFN-FF if you have that available. Right now, if I draw out a borane, I get:
UFF
GFN-FF
GFN1 / GFN2
LJ
If you draw say … benzene or methane, do you at least see UFF?